Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease

 ,
,  ,
, 

{kind=link}
{kind=link}
Abstract
:1. Introduction
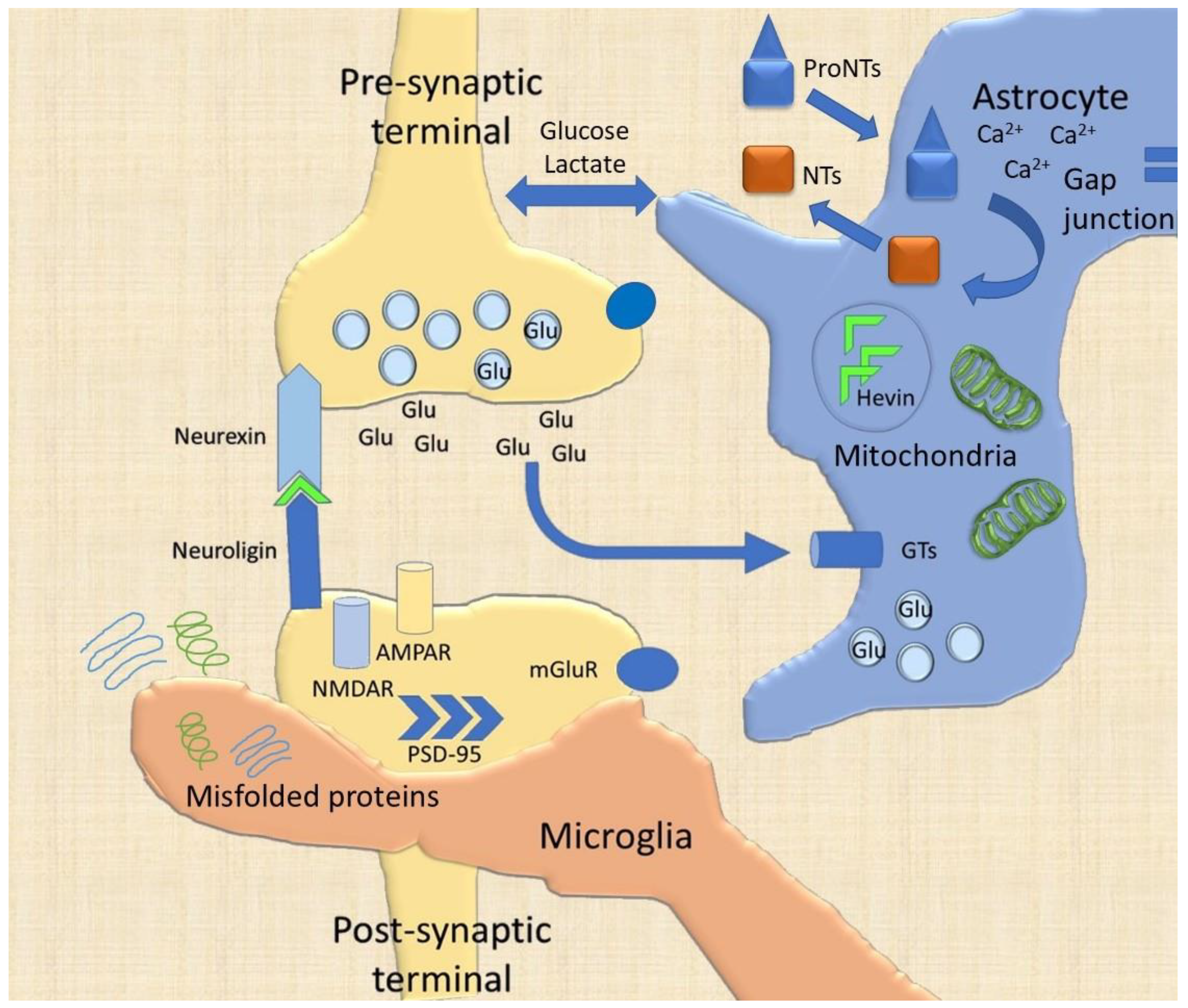
2. The Synaptic Cleft
3. Glial Cells
3.1. Astrocytes
3.2. Microglia
3.3. Oligodendrocytes
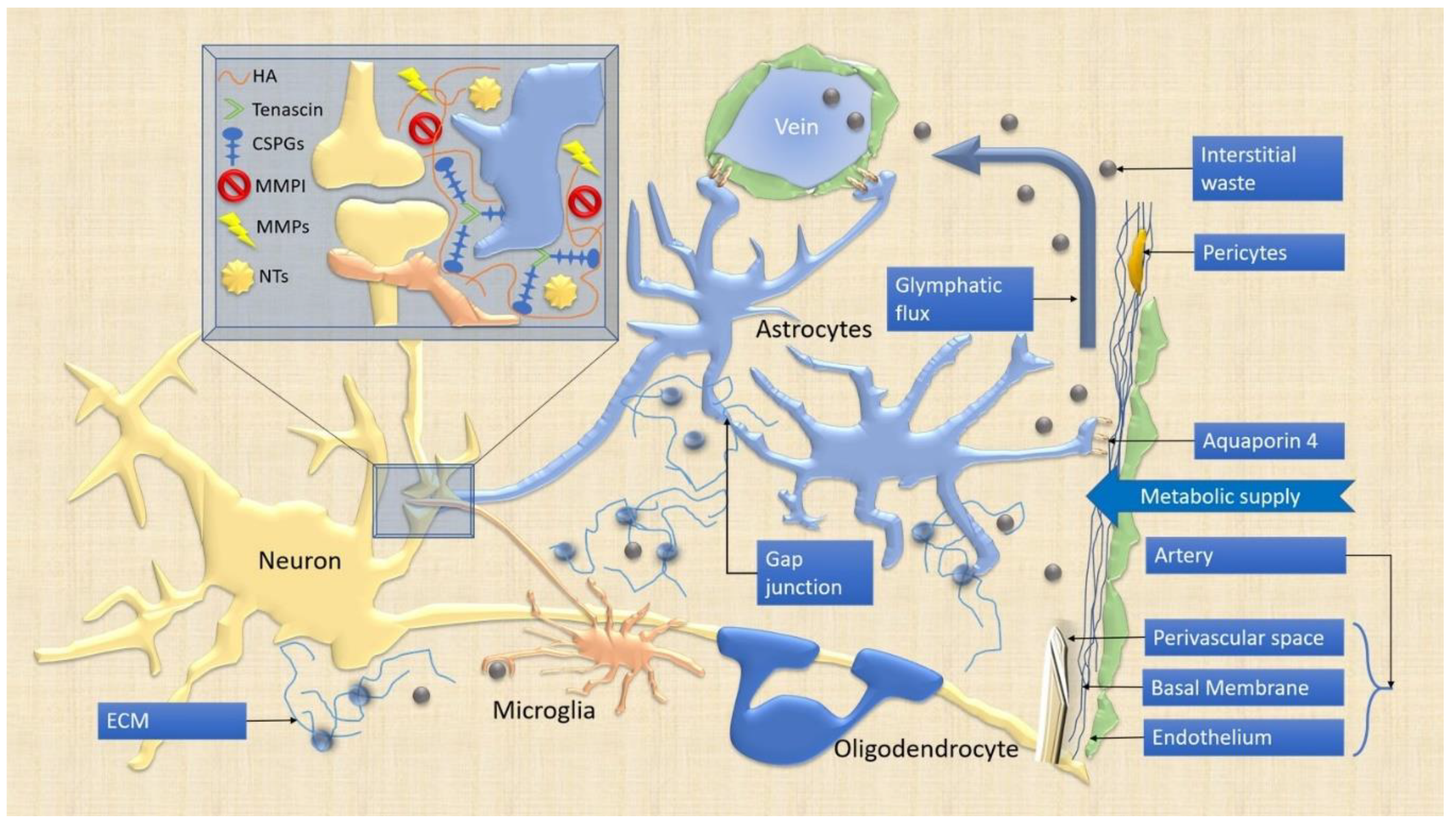
4. ECM and NVU
4.1. ECM
4.2. NVU
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Papa, M.; De Luca, C.; Petta, F.; Alberghina, L.; Cirillo, G. Astrocyte-neuron interplay in maladaptive plasticity. Neurosci. Biobehav. Rev. 2014, 42, 35–54. [Google Scholar] [CrossRef]
- Yamada, S.; Nelson, W.J. Synapses: Sites of cell recognition, adhesion, and functional specification. Annu. Rev. Biochem. 2007, 76, 267–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stogsdill, J.A.; Eroglu, C. The interplay between neurons and glia in synapse development and plasticity. Curr. Opin. Neurobiol. 2017, 42, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, M.; Sellitti, S.; Sadile, A.G. Remodeling of neural networks in the anterior forebrain of an animal model of hyperactivity and attention deficits as monitored by molecular imaging probes. Neurosci. Biobehav. Rev. 2000, 24, 149–156. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Matyash, V.; Kettenmann, H. Heterogeneity in astrocyte morphology and physiology. Brain Res. Rev. 2010, 63, 2–10. [Google Scholar] [CrossRef]
- Farmer, W.T.; Murai, K. Resolving Astrocyte Heterogeneity in the CNS. Front. Cell. Neurosci. 2017, 11, 300. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Richter-Landsberg, C.; Reiser, G. Expression of protease-activated receptors (PARs) in OLN-93 oligodendroglial cells and mechanism of PAR-1-induced calcium signaling. Neuroscience 2004, 126, 69–82. [Google Scholar] [CrossRef]
- Rodríguez-Iglesias, N.; Sierra, A.; Valero, J. Rewiring of Memory Circuits: Connecting Adult Newborn Neurons With the Help of Microglia. Front. Cell Dev. Biol. 2019, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- De Luca, C.; Papa, M. Looking Inside the Matrix: Perineuronal Nets in Plasticity, Maladaptive Plasticity and Neurological Disorders. Neurochem. Res. 2016, 41, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Papa, M. Chapter Five-Matrix Metalloproteinases, Neural Extracellular Matrix, and Central Nervous System Pathology. In Progress in Molecular Biology and Translational Science; Khalil, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 148, pp. 167–202. [Google Scholar]
- Dityatev, A.; Rusakov, D.A. Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 2011, 21, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, C.; Colangelo, A.M.; Alberghina, L.; Papa, M. Neuro-Immune Hemostasis: Homeostasis and Diseases in the Central Nervous System. Front. Cell. Neurosci. 2018, 12, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Davoodi-Bojd, E.; Ding, G.; Zhang, L.; Li, Q.; Li, L.; Chopp, M.; Zhang, Z.; Jiang, Q. Modeling glymphatic system of the brain using MRI. NeuroImage 2019, 188, 616–627. [Google Scholar] [CrossRef]
- Gaglio, D.; Valtorta, S.; Ripamonti, M.; Bonanomi, M.; Damiani, C.; Todde, S.; Negri, A.S.; Sanvito, F.; Mastroianni, F.; Di Campli, A.; et al. Divergent in vitro/in vivo responses to drug treatments of highly aggressive NIH-Ras cancer cells: A PET imaging and metabolomics-mass-spectrometry study. Oncotarget 2016, 7, 52017–52031. [Google Scholar] [CrossRef] [Green Version]
- Calderone, A.; Formenti, M.; Aprea, F.; Papa, M.; Alberghina, L.; Colangelo, A.M.; Bertolazzi, P. Comparing Alzheimer’s and Parkinson’s diseases networks using graph communities structure. BMC Syst. Biol. 2016, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.A.; Ostaszewski, M.; Matsuoka, Y.; Ghosh, S.; Glaab, E.; Trefois, C.; Crespo, I.; Perumal, T.M.; Jurkowski, W.; Antony, P.M.A.; et al. Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol. Neurobiol. 2014, 49, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Colangelo, A.M.; Cirillo, G.; Alberghina, L.; Papa, M.; Westerhoff, H.V. Neural plasticity and adult neurogenesis: The deep biology perspective. Neurotox. Res. 2019, 14, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Foti, M.; Virtuoso, A.; Gaglio, D.; Aprea, F.; Latronico, T.; Rossano, R.; Riccio, P.; Papa, M.; Alberghina, L.; et al. Differential Modulation of NF-κB in Neurons and Astrocytes Underlies Neuroprotection and Antigliosis Activity of Natural Antioxidant Molecules. Oxidative Med. Cell. Longev. 2019, 2019, 8056904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtuoso, A.; Herrera-Rincon, C.; Papa, M.; Panetsos, F. Dependence of Neuroprosthetic Stimulation on the Sensory Modality of the Trigeminal Neurons Following Nerve Injury. Implications in the Design of Future Sensory Neuroprostheses for Correct Perception and Modulation of Neuropathic Pain. Front. Neurosci. 2019, 13, 389. [Google Scholar] [CrossRef]
- Südhof, T.C. The synaptic vesicle cycle: A cascade of protein–protein interactions. Nature 1995, 375, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, F.; Pennuto, M.; Bonanomi, D.; Benfenati, F. Synaptophysin: Leading actor or walk-on role in synaptic vesicle exocytosis? BioEssays 2004, 26, 445–453. [Google Scholar] [CrossRef]
- Südhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227. [Google Scholar] [CrossRef] [PubMed]
- Cupertino, R.B.; Kappel, D.B.; Bandeira, C.E.; Schuch, J.B.; da Silva, B.S.; Müller, D.; Bau, C.H.D.; Mota, N.R. SNARE complex in developmental psychiatry: Neurotransmitter exocytosis and beyond. J. Neural Transm. 2016, 123, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Woodman, P.G. The roles of NSF, SNAPs and SNAREs during membrane fusion. Biochim. Biophys. Acta Mol. Cell Res. 1997, 1357, 155–172. [Google Scholar] [CrossRef] [Green Version]
- Gulsuner, S.; Walsh, T.; Watts, A.C.; Lee, M.K.; Thornton, A.M.; Casadei, S.; Rippey, C.; Shahin, H.; Consortium on the Genetics of Schizophrenia; PAARTNERS Study Group; et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 2013, 154, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landén, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F.; et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef]
- Torres, V.I.; Vallejo, D.; Inestrosa, N.C. Emerging Synaptic Molecules as Candidates in the Etiology of Neurological Disorders. Neural Plast. 2017, 2017, 25. [Google Scholar] [CrossRef]
- Wahlby, C.; Erlandsson, F.; Bengtsson, E.; Zetterberg, A. Sequential immunofluorescence staining and image analysis for detection of large numbers of antigens in individual cell nuclei. Cytometry 2002, 47, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-M.; Veneziano, R.; Gordonov, S.; Li, L.; Danielson, E.; Perez de Arce, K.; Park, D.; Kulesa, A.B.; Wamhoff, E.-C.; Blainey, P.C.; et al. Multiplexed and high-throughput neuronal fluorescence imaging with diffusible probes. Nat. Commun. 2019, 10, 4377. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.L.; Cousin, M.A. The Sybtraps: Control of Synaptobrevin Traffic by Synaptophysin, α-Synuclein and AP-180. Traffic 2014, 15, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, D.; Weisemann, J.; Le Blanc, A.; von Berg, L.; Mahrhold, S.; Piesker, J.; Laue, M.; Luppa, P.B.; Dorner, M.B.; Dorner, B.G.; et al. A lipid-binding loop of botulinum neurotoxin serotypes B, DC and G is an essential feature to confer their exquisite potency. PLOS Pathog. 2018, 14, e1007048. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.M. Bacterial toxins: A table of lethal amounts. Microbiol. Rev. 1982, 46, 86–94. [Google Scholar] [CrossRef]
- Chen, S. Clinical uses of botulinum neurotoxins: Current indications, limitations and future developments. Toxins 2012, 4, 913–939. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-W.; Lee, S.-K.; Ahnn, J. Botulinum Toxin as a Pain Killer: Players and Actions in Antinociception. Toxins 2015, 7, 2435–2453. [Google Scholar] [CrossRef] [Green Version]
- Dini, E.; Mazzucchi, S.; De Luca, C.; Cafalli, M.; Chico, L.; Lo Gerfo, A.; Siciliano, G.; Bonuccelli, U.; Baldacci, F.; Gori, S. Plasma Levels of Oxidative Stress Markers, before and after BoNT/A Treatment, in Chronic Migraine. Toxins 2019, 11, 608. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef] [Green Version]
- Nimchinsky, E.A.; Bernardo, L.S.; Svoboda, K. Structure and Function of Dendritic Spines. Annu. Rev. Physiol. 2002, 64, 313–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckers, T.M. The postsynaptic density. Cell Tissue Res. 2006, 326, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, E. The Postsynaptic Organization of Synapses. Cold Spring Harb. Perspect. Biol. 2011, 3, a005678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Südhof, T.C.; Brose, N. Neuroligins Determine Synapse Maturation and Function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Heine, M.; Thoumine, O.; Mondin, M.; Tessier, B.; Giannone, G.; Choquet, D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc. Natl. Acad. Sci. USA 2008, 105, 20947–20952. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gong, J.; Li, L.; Chen, Y.; Liu, L.; Gu, H.; Luo, X.; Hou, F.; Zhang, J.; Song, R. Neurexin gene family variants as risk factors for autism spectrum disorder. Autism Res. 2018, 11, 37–43. [Google Scholar] [CrossRef]
- Sando, R.; Jiang, X.; Südhof, T.C. Latrophilin GPCRs direct synapse specificity by coincident binding of FLRTs and teneurins. Science 2019, 363, eaav7969. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and Synaptic Plasticity, Cognitive Function, and Dysfunction. In Neurotrophic Factors; Lewin, G.R., Carter, B.D., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 223–250. [Google Scholar]
- Chao, M.V.; Bothwell, M.A.; Ross, A.H.; Koprowski, H.; Lanahan, A.A.; Buck, C.R.; Sehgal, A. Gene transfer and molecular cloning of the human NGF receptor. Science 1986, 232, 518–521. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.; Murinson, B. CHAPTER 25—Therapeutic Potential of Neurotrophic Factors. In From Neuroscience To Neurology; Waxman, S., Ed.; Academic Press: Burlington, NJ, USA, 2005; pp. 419–431. [Google Scholar] [CrossRef]
- Colangelo, A.M.; Cirillo, G.; Lavitrano, M.L.; Alberghina, L.; Papa, M. Targeting reactive astrogliosis by novel biotechnological strategies. Biotechnol. Adv. 2012, 30, 261–271. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Gaglio, D.; Bianco, M.R.; Aprea, F.; Virtuoso, A.; Bonanomi, M.; Alberghina, L.; Papa, M.; Colangelo, A.M. Differentiation by nerve growth factor (NGF) involves mechanisms of crosstalk between energy homeostasis and mitochondrial remodeling. Cell Death Dis. 2018, 9, 391. [Google Scholar] [CrossRef]
- Alberghina, L.; Colangelo, A.M. The modular systems biology approach to investigate the control of apoptosis in Alzheimer’s disease neurodegeneration. BMC Neurosci. 2006, 7, S2. [Google Scholar] [CrossRef] [Green Version]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.-H.; Hortobágyi, T.; Winblad, B.; Ballard, C.; Lehtiö, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: A proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef]
- Witcher, M.R.; Park, Y.D.; Lee, M.R.; Sharma, S.; Harris, K.M.; Kirov, S.A. Three-dimensional relationships between perisynaptic astroglia and human hippocampal synapses. Glia 2010, 58, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Singh, S.K.; Stogsdill, J.A.; Pulimood, N.S.; Dingsdale, H.; Kim, Y.H.; Pilaz, L.-J.; Kim, I.H.; Manhaes, A.C.; Rodrigues, W.S., Jr.; Pamukcu, A.; et al. Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1α and NL1 via Hevin. Cell 2016, 164, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Seifert, G.; Hüttmann, K.; Binder, D.K.; Hartmann, C.; Wyczynski, A.; Neusch, C.; Steinhäuser, C. Analysis of Astroglial K+ Channel Expression in the Developing Hippocampus Reveals a Predominant Role of the Kir4.1 Subunit. J. Neurosci. 2009, 29, 7474. [Google Scholar] [CrossRef]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, B.R.; Assentoft, M.; Cotrina, M.L.; Hua, S.Z.; Nedergaard, M.; Kaila, K.; Voipio, J.; MacAulay, N. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 2014, 62, 608–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrich, G.S.; Kabakov, A.Y.; Hameed, M.Q.; Dhamne, S.C.; Rosenberg, P.A.; Rotenberg, A. Ceftriaxone Treatment after Traumatic Brain Injury Restores Expression of the Glutamate Transporter, GLT-1, Reduces Regional Gliosis, and Reduces Post-Traumatic Seizures in the Rat. J. Neurotrauma 2013, 30, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Lehre, K.P.; Danbolt, N.C. The Number of Glutamate Transporter Subtype Molecules at Glutamatergic Synapses: Chemical and Stereological Quantification in Young Adult Rat Brain. J. Neurosci. 1998, 18, 8751. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.G.; Bardelli, D.; Chiu, M.; Bussolati, O. Changes in the expression of the glutamate transporter EAAT3/EAAC1 in health and disease. Cell. Mol. Life Sci. CMLS 2014, 71, 2001–2015. [Google Scholar] [CrossRef]
- Murphy-Royal, C.; Dupuis, J.P.; Varela, J.A.; Panatier, A.; Pinson, B.; Baufreton, J.; Groc, L.; Oliet, S.H.R. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 2015, 18, 219. [Google Scholar] [CrossRef]
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 13572. [Google Scholar] [CrossRef]
- Yu, X.; Taylor, A.M.W.; Nagai, J.; Golshani, P.; Evans, C.J.; Coppola, G.; Khakh, B.S. Reducing Astrocyte Calcium Signaling In Vivo Alters Striatal Microcircuits and Causes Repetitive Behavior. Neuron 2018, 99, 1170–1187. [Google Scholar] [CrossRef] [Green Version]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Bohmbach, K.; Schwarz, M.K.; Schoch, S.; Henneberger, C. The structural and functional evidence for vesicular release from astrocytes in situ. Brain Res. Bull. 2018, 136, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- De Pittà, M.; Brunel, N. Modulation of Synaptic Plasticity by Glutamatergic Gliotransmission: A Modeling Study. Neural Plast. 2016, 2016, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panatier, A.; Vallée, J.; Haber, M.; Murai, K.K.; Lacaille, J.-C.; Robitaille, R. Astrocytes Are Endogenous Regulators of Basal Transmission at Central Synapses. Cell 2011, 146, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. J. Neurosci. 2002, 22, 183. [Google Scholar] [CrossRef]
- Covelo, A.; Araque, A. Neuronal activity determines distinct gliotransmitter release from a single astrocyte. eLife 2018, 7, e32237. [Google Scholar] [CrossRef]
- Cirillo, G.; Colangelo, A.M.; Berbenni, M.; Ippolito, V.M.; De Luca, C.; Verdesca, F.; Savarese, L.; Alberghina, L.; Maggio, N.; Papa, M. Purinergic Modulation of Spinal Neuroglial Maladaptive Plasticity Following Peripheral Nerve Injury. Mol. Neurobiol. 2015, 52, 1440–1457. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.; Jamison, S.; Robin, L.M.; Zhao, Z.; Martin, E.D.; Aguilar, J.; Benneyworth, M.A.; Marsicano, G.; Araque, A. Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci. 2017, 20, 1540. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gonzalo, M.; Navarrete, M.; Perea, G.; Covelo, A.; Martín-Fernández, M.; Shigemoto, R.; Luján, R.; Araque, A. Endocannabinoids Induce Lateral Long-Term Potentiation of Transmitter Release by Stimulation of Gliotransmission. Cereb. Cortex 2014, 25, 3699–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, L.M.; Oliveira da Cruz, J.F.; Langlais, V.C.; Martin-Fernandez, M.; Metna-Laurent, M.; Busquets-Garcia, A.; Bellocchio, L.; Soria-Gomez, E.; Papouin, T.; Varilh, M.; et al. Astroglial CB1 Receptors Determine Synaptic D-Serine Availability to Enable Recognition Memory. Neuron 2018, 98, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockett, A.T.; Kane, G.A.; Monari, P.K.; Briones, B.A.; Vigneron, P.-A.; Barber, G.A.; Bermudez, A.; Dieffenbach, U.; Kloth, A.D.; Buschman, T.J.; et al. Evidence supporting a role for astrocytes in the regulation of cognitive flexibility and neuronal oscillations through the Ca2+ binding protein S100β. PLoS ONE 2018, 13, e0195726. [Google Scholar] [CrossRef]
- Brown, R.E.; Basheer, R.; McKenna, J.T.; Strecker, R.E.; McCarley, R.W. Control of Sleep and Wakefulness. Physiol. Rev. 2012, 92, 1087–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clasadonte, J.; Scemes, E.; Wang, Z.; Boison, D.; Haydon, P.G. Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron 2017, 95, 1365–1380. [Google Scholar] [CrossRef] [Green Version]
- Poskanzer, K.E.; Yuste, R. Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, E2675. [Google Scholar] [CrossRef] [Green Version]
- Lundgaard, I.; Lu, M.L.; Yang, E.; Peng, W.; Mestre, H.; Hitomi, E.; Deane, R.; Nedergaard, M. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J. Cereb. Blood Flow Metab. 2016, 37, 2112–2124. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science 2013, 342, 373. [Google Scholar] [CrossRef] [Green Version]
- Bacyinski, A.; Xu, M.; Wang, W.; Hu, J. The Paravascular Pathway for Brain Waste Clearance: Current Understanding, Significance and Controversy. Front. Neuroanat. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- De Bock, M.; Leybaert, L.; Giaume, C. Connexin Channels at the Glio-Vascular Interface: Gatekeepers of the Brain. Neurochem. Res. 2017, 42, 2519–2536. [Google Scholar] [CrossRef]
- Pannasch, U.; Freche, D.; Dallerac, G.; Ghezali, G.; Escartin, C.; Ezan, P.; Cohen-Salmon, M.; Benchenane, K.; Abudara, V.; Dufour, A.; et al. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat. Neurosci. 2014, 17, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Vargová, L.; Reingruber, J.; Ezan, P.; Holcman, D.; Giaume, C.; Syková, E.; Rouach, N. Astroglial networks scale synaptic activity and plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 8467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, A.C.; Baca, S.M. Cortical spreading depression and migraine. Nat. Rev. Neurol. 2013, 9, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Hauge, A.W.; Asghar, M.S.; Schytz, H.W.; Christensen, K.; Olesen, J. Effects of tonabersat on migraine with aura: A randomised, double-blind, placebo-controlled crossover study. Lancet Neurol. 2009, 8, 718–723. [Google Scholar] [CrossRef]
- Tarczyluk, M.A.; Nagel, D.A.; O’Neil, J.D.; Parri, H.R.; Tse, E.H.; Coleman, M.D.; Hill, E.J. Functional astrocyte-neuron lactate shuttle in a human stem cell-derived neuronal network. J. Cereb. Blood Flow Metab. 2013, 33, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy Metabolism of the Brain, Including the Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [Green Version]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessieres, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef]
- Haber, M.; Zhou, L.; Murai, K.K. Cooperative Astrocyte and Dendritic Spine Dynamics at Hippocampal Excitatory Synapses. J. Neurosci. 2006, 26, 8881. [Google Scholar] [CrossRef]
- Pirttimaki, T.M.; Hall, S.D.; Parri, H.R. Sustained Neuronal Activity Generated by Glial Plasticity. J. Neurosci. 2011, 31, 7637. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [Green Version]
- Xu-Friedman, M.A.; Harris, K.M.; Regehr, W.G. Three-Dimensional Comparison of Ultrastructural Characteristics at Depressing and Facilitating Synapses onto Cerebellar Purkinje Cells. J. Neurosci. 2001, 21, 6666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, R.; Harris, K.M. Three-Dimensional Relationships between Hippocampal Synapses and Astrocytes. J. Neurosci. 1999, 19, 6897. [Google Scholar] [CrossRef] [PubMed]
- Genoud, C.; Quairiaux, C.; Steiner, P.; Hirling, H.; Welker, E.; Knott, G.W. Plasticity of Astrocytic Coverage and Glutamate Transporter Expression in Adult Mouse Cortex. PLOS Biol. 2006, 4, e343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodosis, D.T.; Poulain, D.A.; Oliet, S.H.R. Activity-Dependent Structural and Functional Plasticity of Astrocyte-Neuron Interactions. Physiol. Rev. 2008, 88, 983–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Cirillo, G.; Colangelo, A.M.; De Luca, C.; Savarese, L.; Barillari, M.R.; Alberghina, L.; Papa, M. Modulation of Matrix Metalloproteinases Activity in the Ventral Horn of the Spinal Cord Re-stores Neuroglial Synaptic Homeostasis and Neurotrophic Support following Peripheral Nerve Injury. PLoS ONE 2016, 11, e0152750. [Google Scholar] [CrossRef]
- De Luca, C.; Savarese, L.; Colangelo, A.M.; Bianco, M.R.; Cirillo, G.; Alberghina, L.; Papa, M. Astrocytes and Microglia-Mediated Immune Response in Maladaptive Plasticity is Differently Modulated by NGF in the Ventral Horn of the Spinal Cord Following Peripheral Nerve Injury. Cell. Mol. Neurobiol. 2016, 36, 37–46. [Google Scholar] [CrossRef]
- Sierra, A.; Beccari, S.; Diaz-Aparicio, I.; Encinas, J.M.; Comeau, S.; Tremblay, M. Surveillance, Phagocytosis, and Inflammation: How Never-Resting Microglia Influence Adult Hippocampal Neurogenesis. Neural Plast. 2014, 2014, 15. [Google Scholar] [CrossRef]
- De Luca, C.; Virtuoso, A.; Maggio, N.; Papa, M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. Int. J. Mol. Sci. 2017, 18, 2128. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841. [Google Scholar] [CrossRef] [Green Version]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röszer, T. Understanding the Biology of Self-Renewing Macrophages. Cells 2018, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, I.; Savage, J.C.; Escobar, L.; Abiega, O.; Gagnon, S.; Hui, C.-W.; Tremblay, M.-È.; Sierra, A.; Valero, J. ProMoIJ: A new tool for automatic three-dimensional analysis of microglial process motility. Glia 2018, 66, 828–845. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Hanamsagar, R.; Bilbo, S.D. Environment matters: Microglia function and dysfunction in a changing world. Curr. Opin. Neurobiol. 2017, 47, 146–155. [Google Scholar] [CrossRef]
- Luo, P.; Chu, S.F.; Zhang, Z.; Xia, C.Y.; Chen, N.H. Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res. Bull. 2019, 146, 12–21. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Roumier, A.; Béchade, C.; Poncer, J.-C.; Smalla, K.-H.; Tomasello, E.; Vivier, E.; Gundelfinger, E.D.; Triller, A.; Bessis, A. Impaired Synaptic Function in the Microglial KARAP/DAP12-Deficient Mouse. J. Neurosci. 2004, 24, 11421. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; Di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef]
- Chen, Z.; Jalabi, W.; Hu, W.; Park, H.-J.; Gale, J.T.; Kidd, G.J.; Bernatowicz, R.; Gossman, Z.C.; Chen, J.T.; Dutta, R.; et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat. Commun. 2014, 5, 4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, K.C.; Josephson, A.; Benusa, S.D.; Hartley, R.K.; Baer, M.; Thummala, S.; Joslyn, M.; Sword, B.A.; Elford, H.; Oh, U.; et al. Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia 2016, 64, 1190–1209. [Google Scholar] [CrossRef] [PubMed]
- Lowery, R.L.; Tremblay, M.-E.; Hopkins, B.E.; Majewska, A.K. The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia 2017, 65, 1744–1761. [Google Scholar] [CrossRef]
- Schecter, R.W.; Maher, E.E.; Welsh, C.A.; Stevens, B.; Erisir, A.; Bear, M.F. Experience-Dependent Synaptic Plasticity in V1 Occurs without Microglial CX3CR1. J. Neurosci. 2017, 37, 10541. [Google Scholar] [CrossRef]
- Ferrini, F.; De Koninck, Y. Microglia Control Neuronal Network Excitability via BDNF Signalling. Neural Plast. 2013, 2013, 11. [Google Scholar] [CrossRef]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, M.; Cumiskey, D.; O’Connor, J.J. Actions of TNF-α on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 2005, 90, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Malik, A.; Choi, H.B.; Ko, R.W.Y.; Dissing-Olesen, L.; MacVicar, B.A. Microglial CR3 Activation Triggers Long-Term Synaptic Depression in the Hippocampus via NADPH Oxidase. Neuron 2014, 82, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- Baron, W.; Colognato, H.; Ffrench-Constant, C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia 2005, 49, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.C.; Bribian, A.; Peregrin, S.; Gil, M.T.; Marin, O.; de Castro, F. Neuregulin-1/ErbB4 signaling controls the migration of oligodendrocyte precursor cells during development. Exp. Neurol. 2012, 235, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Cohen, R.I.; Marmur, R.; Norton, W.T.; Mehler, M.F.; Kessler, J.A. Nerve growth factor and neurotrophin-3 differentially regulate the proliferation and survival of developing rat brain oligodendrocytes. J. Neurosci. 1996, 16, 6433–6442. [Google Scholar] [CrossRef] [Green Version]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Ziskin, J.L.; Nishiyama, A.; Rubio, M.; Fukaya, M.; Bergles, D.E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 2007, 10, 321–330. [Google Scholar] [CrossRef]
- Maldonado, P.P.; Velez-Fort, M.; Angulo, M.C. Is neuronal communication with NG2 cells synaptic or extrasynaptic? J. Anat. 2011, 219, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Balia, M.; Velez-Fort, M.; Passlick, S.; Schafer, C.; Audinat, E.; Steinhauser, C.; Seifert, G.; Angulo, M.C. Postnatal down-regulation of the GABAA receptor gamma2 subunit in neocortical NG2 cells accompanies synaptic-to-extrasynaptic switch in the GABAergic transmission mode. Cereb. Cortex 2015, 25, 1114–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demerens, C.; Stankoff, B.; Logak, M.; Anglade, P.; Allinquant, B.; Couraud, F.; Zalc, B.; Lubetzki, C. Induction of myelination in the central nervous system by electrical activity. Proc. Natl. Acad. Sci. USA 1996, 93, 9887–9892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor skill learning requires active central myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [Green Version]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Sandell, J.H.; Peters, A. Effects of age on the glial cells in the rhesus monkey optic nerve. J. Comp. Neurol. 2002, 445, 13–28. [Google Scholar] [CrossRef]
- Peters, A.; Kemper, T. A review of the structural alterations in the cerebral hemispheres of the aging rhesus monkey. Neurobiol. Aging 2012, 33, 2357–2372. [Google Scholar] [CrossRef] [Green Version]
- Kimura, F.; Itami, C. Myelination and isochronicity in neural networks. Front. Neuroanat. 2009, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Seidl, A.H.; Rubel, E.W.; Barria, A. Differential conduction velocity regulation in ipsilateral and contralateral collaterals innervating brainstem coincidence detector neurons. J. Neurosci. 2014, 34, 4914–4919. [Google Scholar] [CrossRef] [Green Version]
- Sampaio-Baptista, C.; Khrapitchev, A.A.; Foxley, S.; Schlagheck, T.; Scholz, J.; Jbabdi, S.; DeLuca, G.C.; Miller, K.L.; Taylor, A.; Thomas, N.; et al. Motor skill learning induces changes in white matter microstructure and myelination. J. Neurosci. 2013, 33, 19499–19503. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.; Parolisi, R.; Scaroni, F.; Bonfanti, E.; Gualerzi, A.; Gabrielli, M.; Kerlero de Rosbo, N.; Uccelli, A.; Giussani, P.; Viani, P.; et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: Astrocyte involvement in remyelination failure. Acta Neuropathol. 2019, 138, 987–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvetti, L.; Rossi, F. Degradation of Chondroitin Sulfate Proteoglycans Induces Sprouting of Intact Purkinje Axons in the Cerebellum of the Adult Rat. J. Neurosci. 2005, 25, 7150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzwonek, J.; Rylski, M.; Kaczmarek, L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 2004, 567, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Geissler, M.; Gottschling, C.; Aguado, A.; Rauch, U.; Wetzel, C.H.; Hatt, H.; Faissner, A. Primary hippocampal neurons, which lack four crucial extracellular matrix molecules, display abnormalities of synaptic structure and function and severe deficits in perineuronal net formation. J. Neurosci. 2013, 33, 7742–7755. [Google Scholar] [CrossRef] [PubMed]
- Rauch, U.; Zhou, X.H.; Roos, G. Extracellular matrix alterations in brains lacking four of its components. Biochem. Biophys. Res. Commun. 2005, 328, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Saroja, S.R.; Sase, A.; Kircher, S.G.; Wan, J.; Berger, J.; Höger, H.; Pollak, A.; Lubec, G. Hippocampal proteoglycans brevican and versican are linked to spatial memory of Sprague–Dawley rats in the morris water maze. J. Neurochem. 2014, 130, 797–804. [Google Scholar] [CrossRef]
- Milev, P.; Maurel, P.; Chiba, A.; Mevissen, M.; Popp, S.; Yamaguchi, Y.; Margolis, R.K.; Margolis, R.U. Differential Regulation of Expression of Hyaluronan-Binding Proteoglycans in Developing Brain: Aggrecan, Versican, Neurocan, and Brevican. Biochem. Biophys. Res. Commun. 1998, 247, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dino, M.R.; Harroch, S.; Hockfield, S.; Matthews, R.T. Monoclonal antibody Cat-315 detects a glycoform of receptor protein tyrosine phosphatase beta/phosphacan early in CNS development that localizes to extrasynaptic sites prior to synapse formation. Neuroscience 2006, 142, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Gogolla, N.; Caroni, P.; Lüthi, A.; Herry, C. Perineuronal Nets Protect Fear Memories from Erasure. Science 2009, 325, 1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolova, I.; Irintchev, A.; Schachner, M. Tenascin-R restricts posttraumatic remodeling of motoneuron innervation and functional recovery after spinal cord injury in adult mice. J. Neurosci. 2006, 26, 7849–7859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.; Martínez-Sáez, E.; Gutiérrez-Franco, A.; Eixarch, H.; Castro, Z.; Ortega-Aznar, A.; Ramón y Cajal, S.; Montalban, X.; Espejo, C. Expression of semaphorin 3A, semaphorin 7A and their receptors in multiple sclerosis lesions. Mult. Scler. J. 2015, 21, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- McKeon, R.J.; Jurynec, M.J.; Buck, C.R. The Chondroitin Sulfate Proteoglycans Neurocan and Phosphacan Are Expressed by Reactive Astrocytes in the Chronic CNS Glial Scar. J. Neurosci. 1999, 19, 10778. [Google Scholar] [CrossRef]
- Bovolental, P.; Fernaud-Espinosa, I.; Mendez-Otero, R.; Nieto-Sampedro, M. Neurite Outgrowth Inhibitor of Gliotic Brain Tissue. Mode of Action and Cellular Localization, Studied with Specific Monoclonal Antibodies. Eur. J. Neurosci. 1997, 9, 977–989. [Google Scholar] [CrossRef]
- Howell, M.D.; Bailey, L.A.; Cozart, M.A.; Gannon, B.M.; Gottschall, P.E. Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol. Commun. 2015, 3, 54. [Google Scholar] [CrossRef]
- Lessmann, V.; Brigadski, T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: An update. Neurosci. Res. 2009, 65, 11–22. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Inoue, N.; Morita, S.; Nikaido, Y.; Nakashima, T.; Nagai, N.; Okada, K.; Matsuo, O.; Miyata, S. Localization of plasminogen in mouse hippocampus, cerebral cortex, and hypothalamus. Cell Tissue Res. 2011, 343, 303–317. [Google Scholar] [CrossRef]
- Frei, J.A.; Stoeckli, E.T. SynCAMs—From axon guidance to neurodevelopmental disorders. Mol. Cell. Neurosci. 2017, 81, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, T.J.; Tari, P.K.; Connor, S.A.; Zhang, P.; Dobie, F.A.; She, K.; Kawabe, H.; Wang, Y.T.; Brose, N.; Craig, A.M. An LRRTM4-HSPG complex mediates excitatory synapse development on dentate gyrus granule cells. Neuron 2013, 79, 680–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roppongi, R.T.; Karimi, B.; Siddiqui, T.J. Role of LRRTMs in synapse development and plasticity. Neurosci. Res. 2017, 116, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Bhouri, M.; Morishita, W.; Temkin, P.; Goswami, D.; Kawabe, H.; Brose, N.; Sudhof, T.C.; Craig, A.M.; Siddiqui, T.J.; Malenka, R. Deletion of LRRTM1 and LRRTM2 in adult mice impairs basal AMPA receptor transmission and LTP in hippocampal CA1 pyramidal neurons. Proc. Natl. Acad. Sci. USA 2018, 115, E5382–E5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.; Soler-Llavina, G.J.; Fuccillo, M.V.; Malenka, R.C.; Sudhof, T.C. Neuroligins/LRRTMs prevent activity- and Ca2+/calmodulin-dependent synapse elimination in cultured neurons. J. Cell Biol. 2011, 194, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Di Cristo, G.; Chattopadhyaya, B.; Kuhlman, S.J.; Fu, Y.; Belanger, M.C.; Wu, C.Z.; Rutishauser, U.; Maffei, L.; Huang, Z.J. Activity-dependent PSA expression regulates inhibitory maturation and onset of critical period plasticity. Nat. Neurosci. 2007, 10, 1569–1577. [Google Scholar] [CrossRef]
- Muhlenhoff, M.; Rollenhagen, M.; Werneburg, S.; Gerardy-Schahn, R.; Hildebrandt, H. Polysialic acid: Versatile modification of NCAM, SynCAM 1 and neuropilin-2. Neurochem. Res. 2013, 38, 1134–1143. [Google Scholar] [CrossRef]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit—Concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Bartanusz, V.; Jezova, D.; Alajajian, B.; Digicaylioglu, M. The blood-spinal cord barrier: Morphology and Clinical Implications. Ann. Neurol. 2011, 70, 194–206. [Google Scholar] [CrossRef]
- Lampron, A.; ElAli, A.; Rivest, S. Innate Immunity in the CNS: Redefining the Relationship between the CNS and Its Environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoui, Y.; Neal, J.W.; Gasque, P. The Neuro-Immune-Regulators (NIREGs) Promote Tissue Resilience; a Vital Component of the Host’s Defense Strategy against Neuroinflammation. J. Neuroimmune Pharmacol. 2018, 13, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Lee, I.T.; Wu, W.-B.; Liu, C.-J.; Hsieh, H.-L.; Hsiao, L.-D.; Yang, C.-C.; Yang, C.-M. Thrombin Mediates Migration of Rat Brain Astrocytes via PLC, Ca2+, CaMKII, PKCα, and AP-1-Dependent Matrix Metalloproteinase-9 Expression. Mol. Neurobiol. 2013, 48, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Trejo, J. Transactivation of the PAR1-PAR2 Heterodimer by Thrombin Elicits β-Arrestin-mediated Endosomal Signaling. J. Biol. Chem. 2013, 288, 11203–11215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junge, C.E.; Lee, C.J.; Hubbard, K.B.; Zhang, Z.; Olson, J.J.; Hepler, J.R.; Brat, D.J.; Traynelis, S.F. Protease-activated receptor-1 in human brain: Localization and functional expression in astrocytes. Exp. Neurol. 2004, 188, 94–103. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; De Ceunynck, K. Targeting PAR1: Now What? Trends Pharmacol. Sci. 2017, 38, 701–716. [Google Scholar] [CrossRef]
- Rajput, P.S.; Lyden, P.D.; Chen, B.; Lamb, J.A.; Pereira, B.; Lamb, A.; Zhao, L.; Lei, I.F.; Bai, J. Protease activated receptor-1 mediates cytotoxicity during ischemia using in vivo and in vitro models. Neuroscience 2014, 281, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Soh, U.J.K.; Trejo, J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. JTH 2005, 3, 1800–1814. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ricklin, D.; Lambris, J.D. Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc. Natl. Acad. Sci. USA 2017, 114, 10948–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niego, B.; Samson, A.L.; Petersen, K.U.; Medcalf, R.L. Thrombin-induced activation of astrocytes in mixed rat hippocampal cultures is inhibited by soluble thrombomodulin. Brain Res. 2011, 1381, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Shavit Stein, E.; Ben Shimon, M.; Artan Furman, A.; Golderman, V.; Chapman, J.; Maggio, N. Thrombin Inhibition Reduces the Expression of Brain Inflammation Markers upon Systemic LPS Treatment. Neural Plast. 2018, 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padel, T.; Roth, M.; Gaceb, A.; Li, J.Y.; Bjorkqvist, M.; Paul, G. Brain pericyte activation occurs early in Huntington’s disease. Exp. Neurol. 2018, 305, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Xie, L.; Yu, M.; Kang, H.; Feng, T.; Deane, R.; Logan, J.; Nedergaard, M.; Benveniste, H. The Effect of Body Posture on Brain Glymphatic Transport. J. Neurosci. 2015, 35, 11034–11044. [Google Scholar] [CrossRef] [PubMed]
- Petta, F.; De Luca, C.; Triggiani, M.; Casolaro, V. Fragments of truth: T-cell targets of polyclonal immunoglobulins in autoimmune diseases. Curr. Opin. Pharmacol. 2014, 17, 1–11. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, C.; Colangelo, A.M.; Virtuoso, A.; Alberghina, L.; Papa, M. Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1539. https://doi.org/10.3390/ijms21041539
De Luca C, Colangelo AM, Virtuoso A, Alberghina L, Papa M. Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease. International Journal of Molecular Sciences. 2020; 21(4):1539. https://doi.org/10.3390/ijms21041539
Chicago/Turabian StyleDe Luca, Ciro, Anna Maria Colangelo, Assunta Virtuoso, Lilia Alberghina, and Michele Papa. 2020. "Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease" International Journal of Molecular Sciences 21, no. 4: 1539. https://doi.org/10.3390/ijms21041539
APA StyleDe Luca, C., Colangelo, A. M., Virtuoso, A., Alberghina, L., & Papa, M. (2020). Neurons, Glia, Extracellular Matrix and Neurovascular Unit: A Systems Biology Approach to the Complexity of Synaptic Plasticity in Health and Disease. International Journal of Molecular Sciences, 21(4), 1539. https://doi.org/10.3390/ijms21041539