Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity


{kind=link}
Abstract
:1. Introduction
2. NMDAR Structure: Focus on the GluN2A Subunit
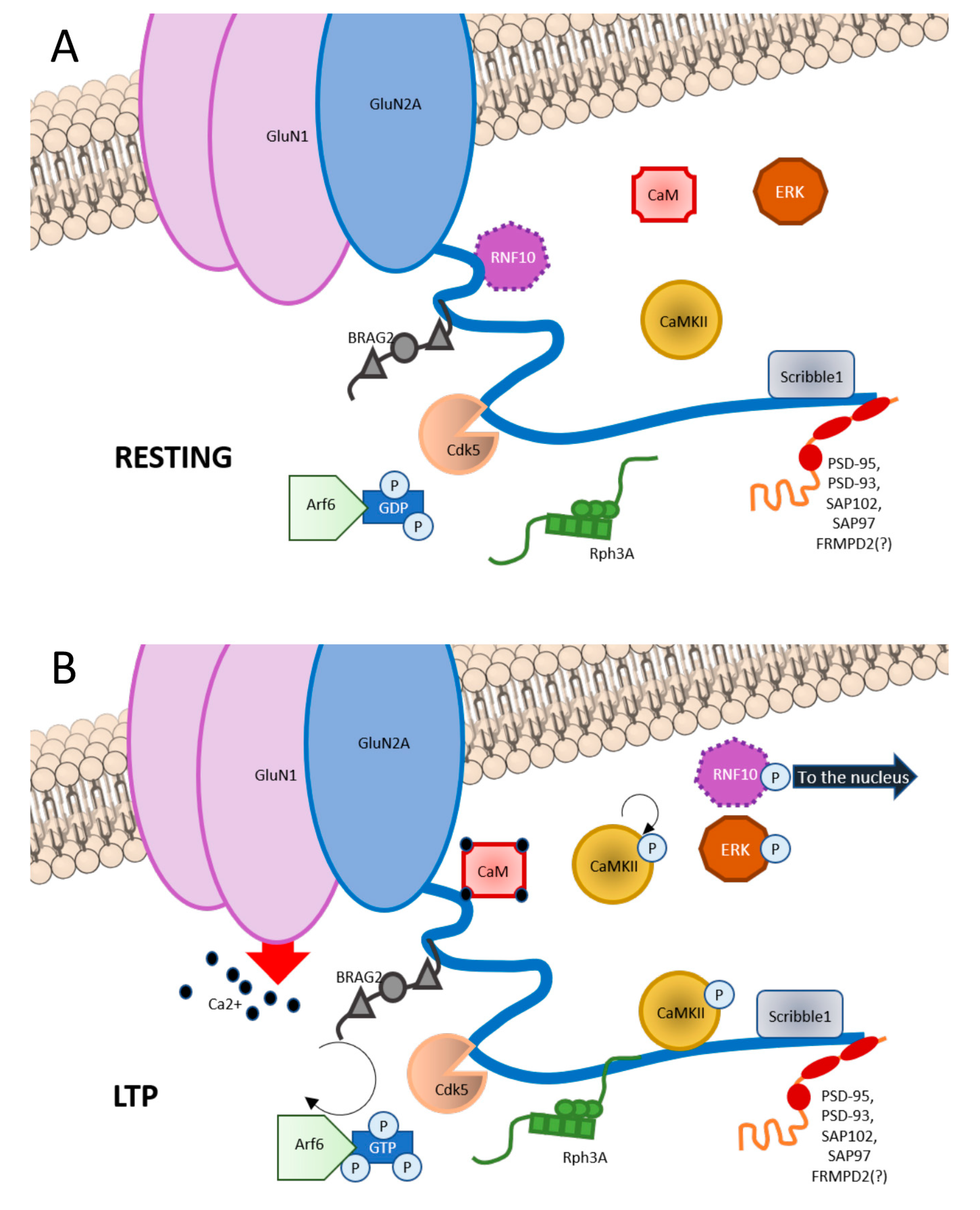
3. GluN2A Subunit: Binding Partners
3.1. Scaffolding Proteins
3.2. Synapse-to-Nucleus Messengers
3.3. Protein Kinases
3.4. Other Proteins
4. GluN2A Subunit and Synaptic Plasticity
4.1. Role of GluN2A in LTP
4.2. Role of GluN2A in LTD
4.3. Role of GluN2A Interactors in Synaptic Plasticity
5. Role of GluN2A in Learning and Memory
6. Role of GluN2A in Pathological Plasticity
6.1. GluN2A in Epilepsy
6.2. GluN2A in Autism Spectrum Disorder (ASD) and in Fragile X Syndrome (FXS)
6.3. Pathological Synaptic Plasticity in Parkinson’s Disease and Dystonia
6.4. Dysfunction of Glutamatergic Synaptic Plasticity in Alzheimer’s Disease
7. Conclusions
Conflicts of Interest
References
- Seeburg, P.H.; Burnashev, N.; Köhr, G.; Kuner, T.; Sprengel, R.; Monyer, H. The NMDA Receptor Channel: Molecular Design of a Coincidence Detector. In Proceedings of the 1993 Laurentian Hormone Conference; Recent progress in hormone research; Academic Press: London, UK, 1995; pp. 19–34. [Google Scholar]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg 2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Duguid, I.; Sjöström, P.J. Novel presynaptic mechanisms for coincidence detection in synaptic plasticity. Curr. Opin. Neurobiol. 2006, 16, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Berretta, N.; Jones, R.S.G. Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience 1996, 75, 339–344. [Google Scholar] [CrossRef]
- Sjöström, P.J.; Turrigiano, G.G.; Nelson, S.B. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 2003, 39, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Siegel, S.J.; Brose, N.; Janssen, W.G.; Gasic, G.P.; Jahn, R.; Heinemann, S.F.; Morrison, J.H. Regional, cellular, and ultrastructural distribution of N-methyl-D- aspartate receptor subunit 1 in monkey hippocampus. Proc. Natl. Acad. Sci. USA 1994, 91, 564–568. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, L.; Taylor, C.; Taylor, R.D.T.; Yau, C.; Langenhan, T.; Hart, M.L.; Christian, H.; Tynan, P.W.; Donnelly, P.; Emptage, N.J. Presynaptic NMDARs in the Hippocampus Facilitate Transmitter Release at Theta Frequency. Neuron 2010, 68, 1109–1127. [Google Scholar] [CrossRef] [Green Version]
- Casado, M.; Dieudonné, S.; Ascher, P. Presynaptic N-methyl-D-aspartate receptors at the parallel fiber-Purkinje cell synapse. Proc. Natl. Acad. Sci. USA 2000, 97, 11593–11597. [Google Scholar] [CrossRef] [Green Version]
- Bidoret, C.; Ayon, A.; Barbour, B.; Casado, M. Presynaptic NR2A-containing NMDA receptors implement a high-pass filter synaptic plasticity rule. Proc. Natl. Acad. Sci. USA 2009, 106, 14126–14131. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, H.; Sheng, M.; Jan, L.Y.; Jan, Y.N.; Basbaum, A.I. Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc. Natl. Acad. Sci. USA 1994, 91, 8383–8387. [Google Scholar] [CrossRef] [Green Version]
- Vissel, B.; Krupp, J.J.; Heinemann, S.F.; Westbrook, G.L. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat. Neurosci. 2001, 4, 587–596. [Google Scholar] [CrossRef]
- Barria, A.; Malinow, R. Subunit-Specific NMDA Receptor Trafficking to Synapses. Neuron 2002, 35, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Nong, Y.; Huang, Y.Q.; Ju, W.; Kalia, L.V.; Ahmadian, G.; Wang, Y.T.; Salter, M.W. Glycine binding primes NMDA receptor internalization. Nature 2003, 422, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.S.; Papouin, T.; Ladépêche, L.; Yao, A.; Langlais, V.C.; Bouchet, D.; Dulong, J.; Mothet, J.P.; Sacchi, S.; Pollegioni, L.; et al. Co-agonists differentially tune GluN2B-NMDA receptor trafficking at hippocampal synapses. Elife 2017, 6, e25492. [Google Scholar] [CrossRef] [PubMed]
- Mayford, M.; Wang, J.; Kandel, E.R.; O’Dell, T.J. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 1995, 81, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Scanziani, M.; Malenka, R.C.; Nicoll, R.A. Role of intercellular interactions in heterosynaptic long-term depression. Nature 1996, 380, 446–450. [Google Scholar] [CrossRef]
- Carter, B.C.; Jahr, C.E. Postsynaptic, not presynaptic NMDA receptors are required for spike-timing-dependent LTD induction. Nat. Neurosci. 2016, 19, 1218–1224. [Google Scholar] [CrossRef]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef] [Green Version]
- Dore, K.; Aow, J.; Malinow, R. Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc. Natl. Acad. Sci. USA 2015, 112, 14705–14710. [Google Scholar] [CrossRef] [Green Version]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Akazawa, C.; Shigemoto, R.; Bessho, Y.; Nakanishi, S.; Mizuno, N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Wu, W.H.; Chen, B.S. NMDA receptor signaling: Death or survival? Front. Biol. (Beijing) 2011, 6, 468–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cercato, M.; Vàzquez, C.; Kornisiuk, E.; Aquirre, A.; Colettis, N.; Snitcofsky, M.; Jerusalinski, D.; Baez, M. GluN1 and GluN2A NMDA Receptor Subunits Increase in the Hippocampus during Memory Consolidation in the Rat. Front. Behav. Neurosci. 2017, 13, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groc, L.; Heine, M.; Cousins, S.L.; Stephenson, F.A.; Lounis, B.; Cognet, L.; Choquet, D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc. Natl. Acad. Sci. USA 2006, 103, 18769–18774. [Google Scholar] [CrossRef] [Green Version]
- Retchless, B.S.; Gao, W.; Johnson, J.W. A single GluN2 subunit residue controls NMDA receptor channel properties via intersubunit interaction. Nat. Neurosci. 2012, 15, 406-S2. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Luo, T.; Raymond, L.A. Subtype-dependence of NMDA receptor channel open probability. J. Neurosci. 1999. [Google Scholar] [CrossRef] [Green Version]
- Flint, A.C.; Maisch, U.S.; Weishaupt, J.H.; Kriegstein, A.R.; Monyer, H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J. Neurosci. 1997. [Google Scholar] [CrossRef]
- Krupp, J.J.; Vissel, B.; Heinemann, S.F.; Westbrook, G.L. Calcium-dependent inactivation of recombinant N-methyl-D-aspartate receptors is NR2 subunit specific. Mol. Pharmacol. 1996, 50, 1680–1688. [Google Scholar]
- Vicini, S.; Wang, J.F.; Li, J.H.; Zhu, W.J.; Wang, Y.H.; Luo, J.H.; Wolfe, B.B.; Grayson, D.R. Functional and pharmacological differences between recombinant N- methyl-D-aspartate receptors. J. Neurophysiol. 1998, 79, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.S.; Hardingham, G.E. The influence of synaptic activity on neuronal health. Curr. Opin. Neurobiol. 2011, 21, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G. NMDA receptor C-terminal signaling in development, plasticity, and disease. F1000Research 2019, 8, 1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Cheng, X.; Zhang, L.; Hu, J.; Chen, Y.; Zhan, L.; Gao, Z. The Functional and Molecular Properties, Physiological Functions, and Pathophysiological Roles of GluN2A in the Central Nervous System. Mol. Neurobiol. 2017, 54, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Baez, M.V.; Cercato, M.C.; Jerusalinsky, D.A. NMDA receptor subunits change after synaptic plasticity induction and learning and memory acquisition. Neural Plast. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Di Luca, M. Targeting glutamatergic synapses in Parkinson’s disease. Curr. Opin. Pharmacol. 2015, 20, 24–28. [Google Scholar] [CrossRef]
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [Google Scholar] [CrossRef]
- Christine, C.W.; Choi, D.W. Effect of zinc on NMDA receptor-mediated channel currents in cortical neurons. J. Neurosci. 1990, 10, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Legendre, P.; Westbrook, G.L. The inhibition of single N-methyl-D-aspartate-activated channels by zinc ions on cultured rat neurones. J. Physiol. 1990, 429, 429–449. [Google Scholar] [CrossRef] [Green Version]
- Fayyazuddin, A.; Villarroel, A.; Le Goff, A.; Lerma, J.; Neyton, J. Four residues of the extracellular N-terminal domain of the NR2A subunit control high-affinity Zn2+ binding to NMDA receptors. Neuron 2000, 25, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Moshaver, A.; Raymond, L.A. Differential sensitivity of recombinant N-methyl-D-aspartate receptor subtypes to zinc inhibition. Mol. Pharmacol. 1997, 51, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.L.; Vyklicky, L. The action of zinc on synaptic transmission and neuronal excitability in cultures of mouse hippocampus. J. Physiol. 1989, 415, 315–365. [Google Scholar] [CrossRef]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 1997, 17, 5711–5725. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Mason-Parker, S.E.; Abraham, W.C.; Tate, W.P. Biphasic changes in the levels of N-methyl-D-aspartate receptor-2 subunits correlate with the induction and persistence of long-term potentiation. Mol. Brain Res. 1998, 60, 21–27. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Cull-Candy, S.G. Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J. Physiol. 1991, 443, 727–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynelis, S.F.; Cull-Candy, S.G. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 1990, 345, 347–350. [Google Scholar] [CrossRef]
- Velíšek, L.; Dreier, J.P.; Stanton, P.K.; Heinemann, U.; Moshé, S.L. Lowering of extracellular pH suppresses low-Mg2+-induces seizures in combined entorhinal cortex-hippocampal slices. Exp. Brain Res. 1994, 101, 44–52. [Google Scholar] [CrossRef]
- Banke, T.G.; Dravid, S.M.; Traynelis, S.F. Protons trap NR1/NR2B NMDA receptors in a nonconducting state. J. Neurosci. 2005, 25, 42–51. [Google Scholar] [CrossRef]
- Zhang, J.B.; Chang, S.; Xu, P.; Miao, M.; Wu, H.; Zhang, Y.; Zhang, T.; Wang, H.; Zhang, J.; Xie, C.; et al. Structural Basis of the Proton Sensitivity of Human GluN1-GluN2A NMDA Receptors. Cell Rep. 2018, 25, 3582–3590. [Google Scholar] [CrossRef] [Green Version]
- Serraz, B.; Grand, T.; Paoletti, P. Altered zinc sensitivity of NMDA receptors harboring clinically-relevant mutations. Neuropharmacology 2016, 109, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Zhang, X.M.; Cao, J.Y.; Yang, W.; Yan, Y.G.; Shan, L.; Zheng, J.; Luo, J.H. An endoplasmic reticulum retention signal located in the extracellular amino-terminal domain of the NR2A subunit of N-methyl-D-aspartate receptors. J. Biol. Chem. 2009, 284, 20285–20298. [Google Scholar] [CrossRef] [Green Version]
- Wollmuth, L.P.; Kuner, T.; Sakmann, B. Adjacent asparagines in the NR2-subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg 2+. J. Physiol. 1998, 506, 13–32. [Google Scholar] [CrossRef]
- Marwick, K.F.M.; Skehel, P.A.; Hardingham, G.E.; Wyllie, D.J.A. The human NMDA receptor GluN2AN615K variant influences channel blocker potency. Pharmacol. Res. Perspect. 2019, 7, e00495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endele, S.; Rosenberger, G.; Geider, K.; Popp, B.; Tamer, C.; Stefanova, I.; Milh, M.; Kortüm, F.; Fritsch, A.; Pientka, F.K.; et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010, 42, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Xu, Y.; Cheng, X.; Chen, X.; Xie, Y.; Zhang, L.; Wang, L.; Hu, J.; Gao, Z. The differences between GluN2A and GluN2B signaling in the brain. J. Neurosci. Res. 2018, 96, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, L.M.; Prybylowski, K.; Chang, K.; Moussan, C.; Stephenson, F.A.; Wenthold, R.J. Export from the endoplasmic reticulum of assembled N-methyl-D-aspartic acid receptors is controlled by a motif in the C terminus of the NR2 subunit. J. Biol. Chem. 2004, 279, 28903–28910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.S.; Roche, K.W. Regulation of NMDA receptors by phosphorylation. Neuropharmacology 2007, 53, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.M.; Philpot, B.D.; Huber, K.M.; Dong, X.; Fallon, J.R.; Bear, M.F. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 2001, 4, 1079–1085. [Google Scholar] [CrossRef]
- Li, B.; Chen, N.; Luo, T.; Otsu, Y.; Murphy, T.H.; Raymond, L.A. Differential regulation of synaptic and extrasynaptic NMDA receptors. Nat. Neurosci. 2002, 5, 833–834. [Google Scholar] [CrossRef]
- Romero-Hernandez, A.; Furukawa, H. Novel mode of antagonist binding in NMDA receptors revealed by the crystal structure of the GluN1-GluN2A ligand-binding domain complexed to NVP-AAM077. Mol. Pharmacol. 2017, 92, 22–29. [Google Scholar] [CrossRef]
- Frizelle, P.; Chen, P.; Wyllie, D. Equilibrium constants for (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) acting at recombinant NR1NR2A and NR1NR2B N-methyl-D-aspartate receptors Im. Mol. Pharmacol. 2006, 70, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, J.A.; Müller, S.L.; Westphälinger, S.E.; Schepmann, D.; Strutz-Seebohm, N.; Seebohm, G.; Wünsch, B. Systematic variation of the benzoylhydrazine moiety of the GluN2A selective NMDA receptor antagonist TCN-201. Eur. J. Med. Chem. 2018, 158, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Mou, T.C.; Dorsett, K.N.; Volkmann, R.A.; Menniti, F.S.; Sprang, S.R.; Hansen, K.B. Structural Basis for Negative Allosteric Modulation of GluN2A-Containing NMDA Receptors. Neuron 2016, 91, 1316–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackos, D.H.; Lupardus, P.J.; Grand, T.; Chen, Y.; Wang, T.M.; Reynen, P.; Gustafson, A.; Wallweber, H.J.A.; Volgraf, M.; Sellers, B.D.; et al. Positive Allosteric Modulators of GluN2A-Containing NMDARs with Distinct Modes of Action and Impacts on Circuit Function. Neuron 2016, 89, 983–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volgraf, M.; Sellers, B.; Jiang, Y.; Wu, G.; Ly, C.; Villemure, E.; Pastor, R.; Yuen, P.; Lu, A.; Luo, X.; et al. Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs) Tuning Deactivation Kinetics via Structure-Based Design. J. Med. Chem. 2016, 59, 2760–2779. [Google Scholar] [CrossRef] [PubMed]
- Villemure, E.; Volgraf, M.; Jiang, Y.; Wu, G.; Ly, C.Q.; Yuen, P.W.; Lu, A.; Luo, X.; Liu, M.; Zhang, S.; et al. GluN2A-selective pyridopyrimidinone series of nmdar positive allosteric modulators with an improved in vivo profile. ACS Med. Chem. Lett. 2016, 8, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, I.A.; Hall, D.D.; Hell, J.W. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J. Biol. Chem. 2002, 277, 21697–21711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornau, H.C.; Schenker, L.T.; Kennedy, M.B.; Seeburg, P.H. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science (80-. ) 1995. [Google Scholar] [CrossRef]
- Howard, M.A.; Elias, G.M.; Elias, L.A.B.; Swat, W.; Nicoll, R.A. The role of SAP97 in synaptic glutamate receptor dynamics. Proc. Natl. Acad. Sci. USA 2010, 107, 3805–3810. [Google Scholar] [CrossRef] [Green Version]
- Sheng, M.; Sala, C. PDZ Domains and the Organization of Supramolecular Complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef]
- Stanic, J.; Carta, M.; Eberini, I.; Pelucchi, S.; Marcello, E.; Genazzani, A.A.; Racca, C.; Mulle, C.; Di Luca, M.; Gardoni, F. Rabphilin 3A retains NMDA receptors at synaptic sites through interaction with GluN2A/PSD-95 complex. Nat. Commun. 2015, 6, 10181. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Zhang, H.; Tian, X.; Wang, X. FRMPD2: A novel GluN2A-interacting scaffold protein in synaptic excitatory transmission. Biochem. Biophys. Res. Commun. 2018. [Google Scholar]
- Piguel, N.H.; Fievre, S.; Blanc, J.M.; Carta, M.; Moreau, M.M.; Moutin, E.; Pinheiro, V.L.; Medina, C.; Ezan, J.; Lasvaux, L.; et al. Scribble1/AP2 complex coordinates NMDA receptor endocytic recycling. Cell Rep. 2014, 9, 712–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panayotis, N.; Karpova, A.; Kreutz, M.R.; Fainzilber, M. Macromolecular transport in synapse to nucleus communication. Trends Neurosci. 2015, 38, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Di Luca, M.; Gardoni, F. Synapse-to-nucleus communication: From developmental disorders to Alzheimer’s disease. Curr. Opin. Neurobiol. 2018, 48, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.C.; Guzzetti, F.; Karpova, A.; Lim, D.; Mitro, N.; Musardo, S.; Mellone, M.; Marcello, E.; Stanic, J.; Samaddar, T.; et al. Ring finger protein 10 is a novel synaptonuclear messenger encoding activation of NMDA receptors in hippocampus. Elife 2016, 5, e12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, B.A.; Fernholz, B.D.; Khatri, L.; Ziff, E.B. Activity-dependent AIDA-1 nuclear signaling regulates nucleolar numbers and protein synthesis in neurons. Nat. Neurosci. 2007. [Google Scholar] [CrossRef]
- Zhai, S.; Ark, E.D.; Parra-Bueno, P.; Yasuda, R. Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science (80-. ) 2013. [Google Scholar] [CrossRef] [Green Version]
- Gardoni, F.; Schrama, L.; Kamal, A.; Gispen, W.; Cattabeni, F.; Di Luca, M. Hippocampal synaptic plasticity involves competition between Ca2+calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J. Neurosci. 2001, 21, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Gardoni, F.; Bellone, C.; Cattabeni, F.; Di Luca, M. Protein Kinase C Activation Modulates α-Calmodulin Kinase II Binding to NR2A Subunit of N-Methyl-d-Aspartate Receptor Complex. J. Biol. Chem. 2001, 273, 20689–20692. [Google Scholar] [CrossRef] [Green Version]
- Strack, S.; Colbran, R.J. Autophosphorylation-dependent targeting of calcium calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 1998, 273, 20689–20692. [Google Scholar] [CrossRef] [Green Version]
- Bayer, K.U.; Schulman, H. Regulation of signal transduction by protein targeting: The case for CaMKII. Biochem. Biophys. Res. Commun. 2001, 289, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.P.; Schulman, H. The Multifunctional Calcium/Calmodulin-Dependent Protein Kinase: From Form to Function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.G.; Kennedy, M.B. Regulation of brain Type II Ca2+ calmodulin-dependent protein kinase by autophosphorylation: A Ca2+-triggered molecular switch. Cell 1986, 44, 861–870. [Google Scholar] [CrossRef]
- Colbran, R.J.; Brown, A.M. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr. Opin. Neurobiol. 2004, 14, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Schrama, L.H.; Van Dalen, J.J.W.; Gispen, W.H.; Cattabeni, F.; Di Luca, M. αCaMKII binding to the C-terminal tail of NMDA receptor subunit NR2A and its modulation by autophosphorylation. FEBS Lett. 1999, 456, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, S.H.; Fu, Y.P.; Wang, J.H.; Lu, Y.M. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat. Neurosci. 2003, 6, 1039–1047. [Google Scholar] [CrossRef]
- Li, B.S.; Sun, M.K.; Zhang, L.; Takahashi, S.; Ma, W.; Vinade, L.; Kulkarni, A.B.; Brady, R.O.; Pant, H.C. Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc. Natl. Acad. Sci. USA 2001, 98, 12742–12747. [Google Scholar] [CrossRef] [Green Version]
- Hisatsune, C.; Umemori, H.; Mishina, M.; Yamamoto, T. Phosphorylation-dependent interaction of the N-methyl-D-aspartate receptor ε2 subunit with phosphatidylinositol 3-kinase. Genes to Cells 1999, 4, 657–666. [Google Scholar] [CrossRef]
- Lee, F.J.S.; Xue, S.; Pei, L.; Vukusic, B.; Chéry, N.; Wang, Y.; Wang, Y.T.; Niznik, H.B.; Yu, X.M.; Liu, F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 2002, 111, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Elagabani, M.N.; Brisevac, D.; Kintscher, M.; Pohle, J.; Köhr, G.; Schmitz, D.; Kornau, H.C. Subunit-selective N-Methyl-D-aspartate (NMDA) receptor signaling through brefeldin a-resistant arf guanine nucleotide exchange factors BRAG1 and BRAG2 during synapse maturation. J. Biol. Chem. 2016, 291, 9105–9118. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, H.; Sanda, M.; Fukaya, M.; Miyazaki, T.; Sukegawa, J.; Yanagisawa, T.; Suzuki, T.; Fukunaga, K.; Watanabe, M.; Kondo, H. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neurosci. Res. 2008, 60, 199–212. [Google Scholar] [CrossRef]
- Li, S.; Tian, X.; Hartley, D.M.; Feig, L.A. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J. Neurosci. 2006, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Gotoh, T.; Tsuji, K.; Lo, E.H.; Huang, S.; Feig, L.A. Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J. 2004, 23, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fam, N.P.; Fan, W.T.; Wang, Z.; Zhang, L.J.; Chen, H.; Moran, M.F. Cloning and characterization of Ras-GRF2, a novel guanine nucleotide exchange factor for Ras. Mol. Cell. Biol. 1997, 17, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shou, C.; Farnsworth, C.L.; Neel, B.G.; Feig, L.A. Molecular cloning of cDNAs encoding a guanine-nucleotide-releasing factor for Ras p21. Nature 1992, 358, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.T.; Koch, C.A.; De Hoog, C.L.; Fam, N.P.; Moran, M.F. The exchange factor Ras-GRF2 activates Ras-dependent and Rac-dependent mitogen-activated protein kinase pathways. Curr. Biol. 1998, 8, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, M.; Zippel, R.; Brambilla, R.; Sturani, E. CDC25(Mm)/Ras-GRF1 regulates both Ras and Rac signaling pathways. FEBS Lett. 1999, 460, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.G.; Li, Z.; Sacks, D.B. IQGAP1 integrates Ca2+calmodulin and B-Raf signaling. J. Biol. Chem. 2008, 283, 22972–22982. [Google Scholar] [CrossRef] [Green Version]
- Reiner, O.; Sapoznik, S.; Sapir, T. Lissencephaly 1 linking to multiple diseases: Mental retardation, neurodegeneration, schizophrenia, male sterility, and more. NeuroMolecular Med. 2006, 8, 547–565. [Google Scholar] [CrossRef]
- Kholmanskikh, S.S.; Koeller, H.B.; Wynshaw-Boris, A.; Gomez, T.; Letourneau, P.C.; Ross, M.E. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat. Neurosci. 2006, 9, 50–57. [Google Scholar] [CrossRef]
- Ryu, J.; Futai, K.; Feliu, M.; Weinberg, R.; Sheng, M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J. Neurosci. 2008, 28, 8178–8188. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, G.; Hau, A.M.; Hsu, P.; Gafken, P.R.; Schimerlik, M.I.; Ishmael, J.E. Identification of an atypical calcium-dependent calmodulin binding site on the C-terminal domain of GluN2A. Biochem. Biophys. Res. Commun. 2014, 44, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Groth, R.D.; Cohen, S.M.; Emery, J.F.; Li, B.; Hoedt, E.; Zhang, G.; Neubert, T.A.; Tsien, R.W. γcaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 2014, 159, 281–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.M.; Suutari, B.; He, X.; Wang, Y.; Sanchez, S.; Tirko, N.N.; Mandelberg, N.J.; Mullins, C.; Zhou, G.; Wang, S.; et al. Calmodulin shuttling mediates cytonuclear signaling to trigger experience-dependent transcription and memory. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.M.; Ma, H.; Kuchibhotla, K.V.; Watson, B.O.; Buzsáki, G.; Froemke, R.C.; Tsien, R.W. Excitation-Transcription Coupling in Parvalbumin-Positive Interneurons Employs a Novel CaM Kinase-Dependent Pathway Distinct from Excitatory Neurons. Neuron 2016, 90, 292–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.M.; Yan, X.Y.; Zhang, B.; Yang, Q.; Ye, M.; Cao, W.; Qiang, W.B.; Zhu, L.J.; Du, Y.L.; Xu, X.X.; et al. Activity-induced synaptic delivery of the GluN2A-containing NMDA receptor is dependent on endoplasmic reticulum chaperone Bip and involved in fear memory. Cell Res. 2015, 25, 818–836. [Google Scholar] [CrossRef] [Green Version]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Li, L.J.; Hu, R.; Lujan, B.; Chen, J.; Zhang, J.J.; Nakano, Y.; Cui, T.Y.; Liao, M.X.; Chen, J.C.; Man, H.Y.; et al. Glycine potentiates AMPA receptor function through metabotropic activation of GluN2A-containing NMDA receptors. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science (80-. ) 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [Green Version]
- Massey, P.V.; Johnson, B.E.; Moult, P.R.; Auberson, Y.P.; Brown, M.W.; Molnar, E.; Collingridge, G.L.; Bashir, Z.I. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 2004, 24, 7821–7828. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Huang, F.S.; Abbas, A.K.; Wigström, H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, T.E.; Bannister, N.J.; Collett, V.J.; Dargan, S.L.; Massey, P.V.; Bortolotto, Z.A.; Fitzjohn, S.M.; Bashir, Z.I.; Collingridge, G.L.; Lodge, D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 2007, 52, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.J.; Russell, K.I.; Wang, Y.T.; Christie, B.R. Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus 2006, 16, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Berberich, S.; Punnakkal, P.; Jensen, V.; Pawlak, V.; Seeburg, P.H.; Hvalby, Ø.; Köhr, G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J. Neurosci. 2005, 25, 6907–6910. [Google Scholar] [CrossRef]
- Romberg, C.; Raffel, J.; Martin, L.; Sprengel, R.; Seeburg, P.H.; Rawlins, J.N.P.; Bannerman, D.M.; Paulsen, O. Induction and expression of GluA1 (GluR-A)-independent LTP in the hippocampus. Eur. J. Neurosci. 2009, 29, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Izumi, Y.; Auberson, Y.P.; Zorumski, C.F. Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J. Neurosci. 2006, 26, 7181–7188. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.; Pitcher, G.M.; Szilard, R.K.; Sertié, A.; Kanisek, M.; Clapcote, S.J.; Lipina, T.; Kalia, L.V.; Joo, D.; McKerlie, C.; et al. Neto1 is a novel CUB-domain NMDA receptor-interacting protein required for synaptic plasticity and learning. PLoS Biol. 2009, 7, e41. [Google Scholar] [CrossRef]
- Sakimura, K.; Kutsuwada, T.; Ito, I.; Manabe, T.; Takayama, C.; Kushiya, E.; Yagi, T.; Shinichi, A.; Inoue, Y.; Sugiyama, H.; et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor 1 subunit. Nature 1995, 373, 151–155. [Google Scholar] [CrossRef]
- Kiyama, Y.; Manabe, T.; Sakimura, K.; Kawakami, F.; Mori, H.; Mishina, M. Increased Thresholds for Long-Term Potentiation and Contextual Learning in Mice Lacking the NMDA-type Glutamate Receptor e1 Subunit. J. Neurosci. 1998, 18, 6704–6712. [Google Scholar] [CrossRef] [Green Version]
- Köhr, G.; Jensen, V.; Koester, H.J.; Mihaljevic, A.L.A.; Utvik, J.K.; Kvello, A.; Ottersen, O.P.; Seeburg, P.H.; Sprengel, R.; Hvalby, Ø. Intracellular Domains of NMDA Receptor Subtypes Are Determinants for Long-Term Potentiation Induction. J. Neurosci. 2003, 23, 10791–10799. [Google Scholar] [CrossRef]
- Moody, T.D.; Watabe, A.M.; Indersmitten, T.; Komiyama, N.H.; Grant, S.G.N.; O’Dell, T.J. β-adrenergic receptor activation rescues theta frequency stimulation-induced LTP deficits in mice expressing C-terminally truncated NMDA receptor GluN2A subunits. Learn. Mem. 2011, 18, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellone, C.; Nicoll, R.A. Rapid Bidirectional Switching of Synaptic NMDA Receptors. Neuron 2007, 55, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Grosshans, D.R.; Clayton, D.A.; Coultrap, S.J.; Browning, M.D. Analysis of glutamate receptor surface expression in acute hippocampal slices. Sci. STKE 2002, 2002, pI8. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.; Stanic, J.; Ponzoni, L.; Mellone, M.; Carrano, N.; Musardo, S.; Zianni, E.; Olivero, G.; Marcello, E.; Pittaluga, A.; et al. Linking NMDA Receptor Synaptic Retention to Synaptic Plasticity and Cognition. iScience 2019, 19, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Baez, M.V.; Oberholzer, M.V.; Cercato, M.C.; Snitcofsky, M.; Aguirre, A.I.; Jerusalinsky, D.A. NMDA Receptor Subunits in the Adult Rat Hippocampus Undergo Similar Changes after 5 Minutes in an Open Field and after LTP Induction. PLoS ONE 2013, 8, e55244. [Google Scholar] [CrossRef] [Green Version]
- Udagawa, T.; Swanger, S.A.; Takeuchi, K.; Kim, J.H.; Nalavadi, V.; Shin, J.; Lorenz, L.J.; Zukin, R.S.; Bassell, G.J.; Richter, J.D. Bidirectional Control of mRNA Translation and Synaptic Plasticity by the Cytoplasmic Polyadenylation Complex. Mol. Cell 2012, 47, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Swanger, S.A.; He, Y.A.; Richter, J.D.; Bassell, G.J. Dendritic GluN2A synthesis mediates activity-induced NMDA receptor insertion. J. Neurosci. 2013, 33, 8898–8908. [Google Scholar] [CrossRef] [Green Version]
- Gerkin, R.C.; Lau, P.-M.; Nauen, D.W.; Wang, Y.T.; Bi, G.-Q. Modular Competition Driven by NMDA Receptor Subtypes in Spike-Timing-Dependent Plasticity. J. Neurophysiol. 2007, 97, 2851–2862. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Dong, Z.; Bagot, R.C.; Howland, J.G.; Phillips, A.G.; Wong, T.P.; Wang, Y.T. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc. Natl. Acad. Sci. USA 2010, 107, 16697–16702. [Google Scholar] [CrossRef] [Green Version]
- Longordo, F.; Kopp, C.; Mishina, M.; Luján, R.; Lüthi, A. NR2A at CA1 synapses is obligatory for the susceptibility of hippocampal plasticity to sleep loss. J. Neurosci. 2009, 29, 9026–9041. [Google Scholar] [CrossRef]
- Cui, Z.; Feng, R.; Jacobs, S.; Duan, Y.; Wang, H.; Cao, X.; Tsien, J.Z. Increased NR2A:NR2B ratio compresses long-term depression range and constrains long-term memory. Sci. Rep. 2013, 3, 1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, T.T.T.; Yang, F.; Chen, B.S.; Lu, Y.; Ji, Y.; Roche, K.W.; Lu, B. Dysbindin regulates hippocampal LTP by controlling NMDA receptor surface expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21395–21400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Carrano, N.; Samaddar, T.; Brunialti, E.; Franchini, L.; Marcello, E.; Ciana, P.; Mauceri, D.; Di Luca, M.; Gardoni, F. The Synaptonuclear Messenger RNF10 Acts as an Architect of Neuronal Morphology. Mol. Neurobiol. 2019, 56, 7583–7593. [Google Scholar] [CrossRef]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 36, 31–39. [Google Scholar] [CrossRef]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G.M. Synaptic Plasticity and Memory: An Evaluation of the Hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.G.M.; Anderson, E.; Lynch, G.S.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 1986, 319, 774–776. [Google Scholar] [CrossRef]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Bannerman, D.M.; Niewoehner, B.; Lyon, L.; Romberg, C.; Schmitt, W.B.; Taylor, A.; Sanderson, D.J.; Cottam, J.; Sprengel, R.; Seeburg, P.H.; et al. NMDA receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J. Neurosci. 2008, 28, 3623–3630. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Liu, S.S.; Yi, F.; Zhuo, M.; Li, B.M. Delay-dependent impairment of spatial working memory with inhibition of NR2B-containing NMDA receptors in hippocampal CA1 region of rats. Mol. Brain 2013. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, M.J.; Abdulla, K.A.; Lindquist, D.H. Neonatal Ethanol Exposure Impairs Trace Fear Conditioning and Alters NMDA Receptor Subunit Expression in Adult Male and Female Rats. Alcohol. Clin. Exp. Res. 2016, 40, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.Q.; Zheng, L.L.; Chen, H.B.; Yu, B.; Wang, W.; Wang, T.; Ruan, B.; Pan, B.X.; Chen, J.R.; Li, X.F.; et al. Hydrogen sulfide reverses aging-associated amygdalar synaptic plasticity and fear memory deficits in rats. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Chen, Z.X.; Wang, R.H.; Shi, Y.W.; Xue, L.; Wang, X.G.; Zhao, H. Knockdown of CLC-3 in the hippocampal CA1 impairs contextual fear memory. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2019, 89, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Cui, Z.; Feng, R.; Wang, H.; Wang, D.; Tsien, J.Z. Molecular and genetic determinants of the NMDA receptor for superior learning and memory functions. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Gupta-Agarwal, S.; Jarome, T.J.; Fernandez, J.; Lubin, F.D. NMDA receptor- and ERK-dependent histone methylation changes in the lateral amygdala bidirectionally regulate fear memory formation. Learn. Mem. 2014, 21, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Holehonnur, R.; Phensy, A.J.; Kim, L.J.; Milivojevic, M.; Vuong, D.; Daison, D.K.; Alex, S.; Tiner, M.; Jones, L.E.; Kroener, S.; et al. Increasing the GluN2A/GluN2B ratio in neurons of the mouse basal and lateral amygdala inhibits the modification of an existing fear memory trace. J. Neurosci. 2016, 36, 9490–9504. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, L.; Gao, Z. Identifying the role of GluN2A in cerebral ischemia. Front. Mol. Neurosci. 2017. [Google Scholar] [CrossRef]
- Boyce-Rustay, J.M.; Holmes, A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology 2006, 31, 2405–2414. [Google Scholar] [CrossRef]
- Taniguchi, S.; Nakazawa, T.; Tanimura, A.; Kiyama, Y.; Tezuka, T.; Watabe, A.M.; Katayama, N.; Yokoyama, K.; Inoue, T.; Izumi-Nakaseko, H.; et al. Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J. 2009, 28, 3717–3729. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Shao, W.; Liu, Z.; Fan, S.; Yu, J.; Chen, J.; Qiao, R.; Zhou, J.; Xie, P. ITRAQ-based quantitative analysis of hippocampal postsynaptic density-associated proteins in a rat chronic mild stress model of depression. Neuroscience 2015, 298, 220–292. [Google Scholar] [CrossRef]
- Feyissa, A.M.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2009, 33, 70–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karolewicz, B.; Szebeni, K.; Gilmore, T.; MacIag, D.; Stockmeier, C.A.; Ordway, G.A. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int. J. Neuropsychopharmacol. 2009, 12, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaut, O.; Schmitt, I.; Hofmann, A.; Hoffmann, P.; Schlaepfer, T.E.; Wüllner, U.; Hurlemann, R. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Francija, E.; Petrovic, Z.; Brkic, Z.; Mitic, M.; Radulovic, J.; Adzic, M. Disruption of the NMDA receptor GluN2A subunit abolishes inflammation-induced depression. Behav. Brain Res. 2019, 359, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Guan, L.; Zhu, Z.; Li, H. Reduced levels of NR1 and NR2A with depression-like behavior in different brain regions in prenatally stressed juvenile offspring. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Jia, N.; Guan, L.; Su, Q.; Wang, D.; Li, H.; Zhu, Z. Involvement of NR1, NR2A different expression in brain regions in anxiety-like behavior of prenatally stressed offspring. Behav. Brain Res. 2013, 257, 1–7. [Google Scholar] [CrossRef]
- Itokawa, M.; Yamada, K.; Yoshitsugu, K.; Toyota, T.; Suga, T.; Ohba, H.; Watanabe, A.; Hattori, E.; Shimizu, H.; Kumakura, T.; et al. A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics 2003, 13, 271–278. [Google Scholar] [CrossRef]
- Pinacho, R.; Villalmanzo, N.; Roca, M.; Iniesta, R.; Monje, A.; Haro, J.M.; Meana, J.J.; Ferrer, I.; Gill, G.; Ramos, B. Analysis of Sp transcription factors in the postmortem brain of chronic schizophrenia: A pilot study of relationship to negative symptoms. J. Psychiatr. Res. 2013, 47, 926–934. [Google Scholar] [CrossRef]
- Beneyto, M.; Meador-Woodruff, J.H. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 2008, 33, 2175–2186. [Google Scholar] [CrossRef]
- Bitanihirwe, B.K.Y.; Lim, M.P.; Kelley, J.F.; Kaneko, T.; Woo, T.U.W. Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 2009, 9. [Google Scholar] [CrossRef] [Green Version]
- Arning, L.; Kraus, P.H.; Valentin, S.; Saft, C.; Andrich, J.; Epplen, J.T. NR2A and NR2B receptor gene variations modify age at onset in Huntington disease. Neurogenetics 2005, 6, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Apostol, B.L.; Dunah, A.W.; DeJohn, M.M.; Farrell, L.A.; Bates, G.P.; Young, A.B.; Standaert, D.G.; Thompson, L.M.; Cha, J.H.J. Complex alteration of NMDA receptors in transgenic Huntington’s disease mouse brain: Analysis of mRNA and protein expression, plasma membrane association, interacting proteins, and phosphorylation. Neurobiol. Dis. 2003, 14, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.J.; Levine, M.S. Changes in expression of N-methyl-D-aspartate receptor subunits occur early in the R6/2 mouse model of Huntington’s disease. Dev. Neurosci. 2006, 28, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Vezzani, A.; O’rien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 3, 18024. [Google Scholar]
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Bruneau, N.; Lozovaya, N.; Labalme, A.; Boutry-Kryza, N.; Salmi, M.; Tsintsadze, T.; Addis, L.; Motte, J.; et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat. Genet. 2013, 45, 1061–1066. [Google Scholar] [CrossRef]
- Carvill, G.L.; Regan, B.M.; Yendle, S.C.; O’Roak, B.J.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- De Ligt, J.; Willemsen, M.H.; Van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; De Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Ogden, K.K.; Chen, W.; Swanger, S.A.; McDaniel, M.J.; Fan, L.Z.; Hu, C.; Tankovic, A.; Kusumoto, H.; Kosobucki, G.J.; Schulien, A.J.; et al. Molecular Mechanism of Disease-Associated Mutations in the Pre-M1 Helix of NMDA Receptors and Potential Rescue Pharmacology. PLoS Genet. 2017, 13, e1006536. [Google Scholar] [CrossRef]
- Pierson, T.M.; Yuan, H.; Marsh, E.D.; Fuentes-Fajardo, K.; Adams, D.R.; Markello, T.; Golas, G.; Simeonov, D.R.; Holloman, C.; Tankovic, A.; et al. GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann. Clin. Transl. Neurol. 2014, 1, 190–198. [Google Scholar] [CrossRef]
- Yuan, H.; Hansen, K.B.; Zhang, J.; Mark Pierson, T.; Markello, T.C.; Fajardo, K.V.F.; Holloman, C.M.; Golas, G.; Adams, D.R.; Boerkoel, C.F.; et al. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat. Commun. 2014, 5, 3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Tankovic, A.; Burger, P.B.; Kusumoto, H.; Traynelis, S.F.; Yuan, H. Functional evaluation of a de novo GRIN2A mutation identified in a patient with profound global developmental delay and refractory epilepsy. Mol. Pharmacol. 2017, 91, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.W.; Kalia, L.V. SRC kinases: A hub for NMDA receptor regulation. Nat. Rev. Neurosci. 2004, 5, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Bruneau, N.; Antonov, S.M.; Szepetowski, P.; Burnashev, N.; Giniatullin, R. Functional properties of human NMDA receptors associated with epilepsy-related mutations of GluN2A subunit. Front. Cell. Neurosci. 2017, 155. [Google Scholar] [CrossRef]
- Lemke, J.R.; Geider, K.; Helbig, K.L.; Heyne, H.O.; Schütz, H.; Hentschel, J.; Courage, C.; Depienne, C.; Nava, C.; Heron, D.; et al. Delineating the GRIN1 phenotypic spectrum: A distinct genetic NMDA receptor encephalopathy. Neurology 2016, 86, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- Swanger, S.A.; Chen, W.; Wells, G.; Burger, P.B.; Tankovic, A.; Bhattacharya, S.; Strong, K.L.; Hu, C.; Kusumoto, H.; Zhang, J.; et al. Mechanistic Insight into NMDA Receptor Dysregulation by Rare Variants in the GluN2A and GluN2B Agonist Binding Domains. Am. J. Hum. Genet. 2016, 99, 1261–1280. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.X.; Luo, J.H. Mutations of N-Methyl-D-Aspartate Receptor Subunits in Epilepsy. Neurosci. Bull. 2018, 34, 549–565. [Google Scholar] [CrossRef]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar]
- Adams, D.R.; Yuan, H.; Holyoak, T.; Arajs, K.H.; Hakimi, P.; Markello, T.C.; Wolfe, L.A.; Vilboux, T.; Burton, B.K.; Fajardo, K.F.; et al. Three rare diseases in one Sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith-Magenis Syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity. Mol. Genet. Metab. 2014, 113, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Gori, M.B.; Girardi, E. 3-Mercaptopropionic acid-induced repetitive seizures increase glun2a expression in rat hippocampus: A potential neuroprotective role of cyclopentyladenosine. Cell. Mol. Neurobiol. 2013, 33, 803–813. [Google Scholar] [CrossRef]
- Borbély, S.; Dobó, E.; Czégé, D.; Molnár, E.; Bakos, M.; Szucs, B.; Vincze, A.; Világi, I.; Mihály, A. Modification of ionotropic glutamate receptor-mediated processes in the rat hippocampus following repeated, brief seizures. Neuroscience 2009, 159, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; He, S.; Hu, X.L.; Yu, J.; Zhou, Y.; Zheng, J.; Zhang, S.; Zhang, C.; Duan, W.H.; Xiong, Z.Q. Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. J. Neurosci. 2007, 27, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tian, X.; Xu, D.; Zheng, F.; Lu, X.; Zhang, Y.; Ma, Y.; Li, Y.; Xu, X.; Zhu, B.; et al. GPR40 modulates epileptic seizure and NMDA receptor function. Sci. Adv. 2018, 4, eaau2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abegg, M.H.; Savic, N.; Ehrengruber, M.U.; McKinney, R.A.; Gähwiler, B.H. Epileptiform activity in rat hippocampus strengthens excitatory synapses. J. Physiol. 2004, 554, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Cain, D.P.; Hargreaves, E.L.; Boon, F.; Dennison, Z. An examination of the relations between hippocampal long-term potentiation, kindling, afterdischarge, and place learning in the water maze. Hippocampus 1993, 3, 153–163. [Google Scholar] [CrossRef]
- McNamara, R.K.; Kirkby, R.D.; DePape, G.E.; Skelton, R.W.; Corcoran, M.E. Differential effects of kindling and kindled seizures on place learning in the morris water maze. Hippocampus 1993, 3, 149–152. [Google Scholar] [CrossRef]
- Rutten, A.; Van Albada, M.; Silveira, D.C.; Cha, B.H.; Liu, X.; Hu, Y.N.; Cilio, M.R.; Holmes, G.L. Memory impairment following status epilepticus in immature rats: Time-course and environmental effects. Eur. J. Neurosci. 2002, 16, 501–513. [Google Scholar] [CrossRef]
- Guli, X.; Tokay, T.; Kirschstein, T.; Köhling, R. Status Epilepticus Enhances Depotentiation after Fully Established LTP in an NMDAR-Dependent but GluN2B-Independent Manner. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, H.; Bernard, P.B.; Castano, A.M.; Benke, T.A. Enhanced long term potentiation and decreased AMPA receptor desensitization in the acute period following a single kainate induced early life seizure. Neurobiol. Dis. 2016, 87, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Tokay, T.; Porath, K.; Köhling, R.; Kirschstein, T. Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 2013, 54, 183–193. [Google Scholar] [CrossRef]
- Kryukov, K.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status epilepticus alters hippocampal long-term synaptic potentiation in a rat lithium-pilocarpine model. Neuroreport 2016, 27, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.L.; Shatskikh, T.N.; Liu, X.; Holmes, G.L. Impaired single cell firing and long-term potentiation parallels memory impairment following recurrent seizures. Eur. J. Neurosci. 2007, 25, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Suárez, L.M.; Cid, E.; Gal, B.; Inostroza, M.; Brotons-Mas, J.R.; Gómez-Domínguez, D.; de la Prida, L.M.; Solís, J.M. Systemic Injection of Kainic Acid Differently Affects LTP Magnitude Depending on its Epileptogenic Efficiency. PLoS ONE 2012, 7, e48128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, J.E.; Ma, K.; Elstrott, J.; Weber, M.; Saillet, S.; Khan, A.S.; Simms, J.; Liu, B.; Kim, T.A.; Yu, G.Q.; et al. GluN2A NMDA Receptor Enhancement Improves Brain Oscillations, Synchrony, and Cognitive Functions in Dravet Syndrome and Alzheimer’s Disease Models. Cell Rep. 2020, 30, 381–396.e4. [Google Scholar] [CrossRef]
- Bagni, C.; Zukin, R.S. A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.; Acharya, U.R.; Adeli, H.; Bairy, G.M.; Adeli, A. Autism: Cause factors, early diagnosis and therapies. Rev. Neurosci. 2014, 25, 841–850. [Google Scholar] [CrossRef]
- Volkmar, F.R.; McPartland, J.C. From Kanner to DSM-5: Autism as an Evolving Diagnostic Concept. Annu. Rev. Clin. Psychol. 2014, 10, 193–212. [Google Scholar] [CrossRef] [Green Version]
- Park, H.R.; Lee, J.M.; Moon, H.E.; Lee, D.S.; Kim, B.N.; Kim, J.; Kim, D.G.; Paek, S.H. A short review on the current understanding of autism spectrum disorders. Exp. Neurobiol. 2016, 25, 1–13. [Google Scholar] [CrossRef]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A Review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef]
- Bagni, C.; Tassone, F.; Neri, G.; Hagerman, R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. J. Clin. Invest. 2012, 122, 4314–4322. [Google Scholar] [CrossRef] [Green Version]
- Bassell, G.J.; Warren, S.T. Fragile X Syndrome: Loss of Local mRNA Regulation Alters Synaptic Development and Function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rubeis, S.; Bagni, C. Fragile X mental retardation protein control of neuronal mRNA metabolism: Insights into mRNA stability. Mol. Cell. Neurosci. 2010, 43, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Bagni, C. SnapShot: FMRP mRNA targets and diseases. Cell 2014, 158, 1446–1446.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicka, K.; Pyronneau, A.; Chao, M.; Bennett, M.V.L.; Zukin, R.S. Elevated erk/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in Fragile X mice. Proc. Natl. Acad. Sci. USA 2016, 113, E6290–E6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Snape, M.; Klann, E.; Stone, J.G.; Singh, A.; Petersen, R.B.; Castellani, R.J.; Casadesus, G.; Smith, M.A.; Zhu, X. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J. Neurochem. 2012, 121, 672–679. [Google Scholar] [CrossRef]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [Green Version]
- Lauterborn, J.C.; Rex, C.S.; Kramár, E.; Chen, L.Y.; Pandyarajan, V.; Lynch, G.; Gall, C.M. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J. Neurosci. 2007, 27, 10685–10694. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Ge, W.P.; Huang, W.; He, Y.; Wang, G.X.; Rowson-Baldwin, A.; Smith, S.J.; Jan, Y.N.; Jan, L.Y. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 2011, 72, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Paradee, W.; Melikian, H.E.; Rasmussen, D.L.; Kenneson, A.; Conn, P.J.; Warren, S.T. Fragile X mouse: Strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 1999, 94, 185–192. [Google Scholar] [CrossRef]
- Zhao, M.G.; Toyoda, H.; Ko, S.W.; Ding, H.K.; Wu, L.J.; Zhuo, M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 2005, 25, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.S.; Casimiro, T.M.; Gruber, S.M.; Vanderklish, P.W. Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 2006, 96, 1734–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meredith, R.M.; Holmgren, C.D.; Weidum, M.; Burnashev, N.; Mansvelder, H.D. Increased Threshold for Spike-Timing-Dependent Plasticity Is Caused by Unreliable Calcium Signaling in Mice Lacking Fragile X Gene Fmr1. Neuron 2007, 54, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Wang, H.; Mercaldo, V.; Li, X.; Chen, T.; Zhuo, M. Fragile X mental retardation protein is required for chemically-induced long-term potentiation of the hippocampus in adult mice. J. Neurochem. 2009, 111, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundbye, C.J.; Toft, A.K.H.; Banke, T.G. Inhibition of GluN2A NMDA receptors ameliorates synaptic plasticity deficits in the Fmr1−/y mouse model. J. Physiol. 2018, 596, 5017–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.H.; Trommer, B.L. Fragile X mice: Reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J. Neurosci. Res. 2011, 89, 176–182. [Google Scholar] [CrossRef]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [Green Version]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- Eadie, B.D.; Cushman, J.; Kannangara, T.S.; Fanselow, M.S.; Christie, B.R. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 2012, 22, 241–254. [Google Scholar] [CrossRef]
- McHugh, T.J.; Jones, M.W.; Quinn, J.J.; Balthasar, N.; Coppari, R.; Elmquist, J.K.; Lowell, B.B.; Fanselow, M.S.; Wilson, M.A.; Tonegawa, S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science (80-. ) 2007, 317, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Eadie, B.D.; Zhang, W.N.; Boehme, F.; Gil-Mohapel, J.; Kainer, L.; Simpson, J.M.; Christie, B.R. Fmr1 knockout mice show reduced anxiety and alterations in neurogenesis that are specific to the ventral dentate gyrus. Neurobiol. Dis. 2009, 36, 361–373. [Google Scholar] [CrossRef] [PubMed]
- D’Hooge, R.; Nagels, G.; Franck, F.; Bakker, C.E.; Reyniers, E.; Storm, K.; Kooy, R.F.; Oostra, B.A.; Willems, P.J.; De Deyn, P.P. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 1997, 76, 367–376. [Google Scholar] [CrossRef]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, G.L.; Wang, Y.T.; Floresco, S.B.; Phillips, A.G. Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- Duan, W.; Wang, K.; Duan, Y.; Chu, X.; Ma, R.; Hu, P.; Xiong, B. Integrated Transcriptome Analyses Revealed Key Target Genes in Mouse Models of Autism. Autism Res. 2019. [Google Scholar] [CrossRef]
- Gardoni, F.; Bellone, C. Modulation of the glutamatergic transmission by Dopamine: A focus on Parkinson, Huntington and Addiction diseases. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Di Filippo, M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007, 30, 211–219. [Google Scholar] [CrossRef]
- Dunah, A.W.; Wang, Y.; Yasuda, R.P.; Kameyama, K.; Huganir, R.L.; Wolfe, B.B.; Standaert, D.G. Alterations in subunit expression, composition, and phosphorylation of striatal N-methyl-D-aspartate glutamate receptors in a rat 6-hydroxydopamine model of Parkinson’s disease. Mol. Pharmacol. 2000, 57, 342–352. [Google Scholar]
- Gardoni, F.; Picconi, B.; Ghiglieri, V.; Polli, F.; Bagetta, V.; Bernardi, G.; Cattabeni, F.; Di Luca, M.; Calabresi, P. A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J. Neurosci. 2006, 26, 2914–2922. [Google Scholar] [CrossRef] [Green Version]
- Paillé, V.; Picconi, B.; Bagetta, V.; Ghiglieri, V.; Sgobio, C.; Di Filippo, M.; Viscomi, M.T.; Giampà, C.; Fusco, F.R.; Gardoni, F.; et al. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J. Neurosci. 2010, 30, 14182–14193. [Google Scholar] [CrossRef]
- Mellone, M.; Stanic, J.; Hernandez, L.F.; Iglesias, E.; Zianni, E.; Longhi, A.; Prigent, A.; Picconi, B.; Calabresi, P.; Hirsch, E.C.; et al. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: From experimental models to patients. Front. Cell. Neurosci. 2015, 9, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, V.; De Iure, A.; Loffredo, V.; Vaikath, N.; De Risi, M.; Paciotti, S.; Quiroga-Varela, A.; Chiasserini, D.; Mellone, M.; Mazzocchetti, P.; et al. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain 2019, 142, 1365–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diógenes, M.J.; Dias, R.B.; Rombo, D.M.; Vicente Miranda, H.; Maiolino, F.; Guerreiro, P.; Näsström, T.; Franquelim, H.G.; Oliveira, L.M.A.; Castanho, M.A.R.B.; et al. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rascol, O. Efficacy and safety of amantadine for the treatment of l-DOPA-induced dyskinesia. J. Neural Transm. 2018, 125, 1237–1250. [Google Scholar] [CrossRef]
- Picconi, B.; Centonze, D.; Håkansson, K.; Bernardi, G.; Greengard, P.; Fisone, G.; Cenci, M.A.; Calabresi, P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 2003, 6, 501–506. [Google Scholar] [CrossRef]
- Prescott, I.A.; Liu, L.D.; Dostrovsky, J.O.; Hodaie, M.; Lozano, A.M.; Hutchison, W.D. Lack of depotentiation at basal ganglia output neurons in PD patients with levodopa-induced dyskinesia. Neurobiol. Dis. 2014, 71, 24–33. [Google Scholar] [CrossRef]
- Hallett, P.J.; Dunah, A.W.; Ravenscroft, P.; Zhou, S.; Bezard, E.; Crossman, A.R.; Brotchie, J.M.; Standaert, D.G. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology 2005, 48, 503–516. [Google Scholar] [CrossRef]
- Gardoni, F.; Sgobio, C.; Pendolino, V.; Calabresi, P.; Di Luca, M.; Picconi, B. Targeting NR2A-containing NMDA receptors reduces L-DOPA-induced dyskinesias. Neurobiol. Aging 2012, 33, 2138–2144. [Google Scholar] [CrossRef]
- Stanic, J.; Mellone, M.; Napolitano, F.; Racca, C.; Zianni, E.; Minocci, D.; Ghiglieri, V.; Thiolat, M.L.; Li, Q.; Longhi, A.; et al. Rabphilin 3A: A novel target for the treatment of levodopa-induced dyskinesias. Neurobiol. Dis. 2017, 108, 54–64. [Google Scholar] [CrossRef]
- Quartarone, A.; Hallett, M. Emerging concepts in the physiological basis of dystonia. Mov. Disord. 2013, 28, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Quartarone, A.; Rizzo, V.; Terranova, C.; Morgante, F.; Schneider, S.; Ibrahim, N.; Girlanda, P.; Bhatia, K.P.; Rothwell, J.C. Abnormal sensorimotor plasticity in organic but not in psychogenic dystonia. Brain 2009, 132, 2871–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltese, M.; Stanic, J.; Tassone, A.; Sciamanna, G.; Ponterio, G.; Vanni, V.; Martella, G.; Imbriani, P.; Bonsi, P.; Mercuri, N.B.; et al. Early structural and functional plasticity alterations in a susceptibility period of DYT1 dystonia mouse striatum. Elife 2018, 7, e33331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Anwyl, R.; Suh, Y.H.; Djamgoz, M.B.A.; Rowan, M.J. Use-dependent effects of amyloidogenic fragments of β-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J. Neurosci. 2001, 21, 1327–1333. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid β Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.W.; Klyubin, I.; Anwy, R.; Rowan, M.J. GluN2B subunit-containing NMDA receptor antagonists prevent Aβ-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20504–20509. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble a β oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Rönicke, R.; Mikhaylova, M.; Rönicke, S.; Meinhardt, J.; Schröder, U.H.; Fändrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef]
- Texidó, L.; Martín-Satué, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Aoki, C.; Lee, J.; Nedelescu, H.; Ahmed, T.; Ho, A.; Shen, J. Increased levels of NMDA receptor NR2A subunits at pre- and postsynaptic sites of the hippocampal CA1: An early response to conditional double knockout of presenilin 1 and 2. J. Comp. Neurol. 2009, 517, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessels, H.W.; Nabavi, S.; Malinow, R. Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4033–4038. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tak, P.W.; Aarts, M.; Rooyakkers, A.; Liu, L.; Ted, W.L.; Dong, C.W.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, E.; Musardo, S.; Vandermeulen, L.; Pelucchi, S.; Gardoni, F.; Santo, N.; Antonucci, F.; Di Luca, M. Amyloid-β Oligomers Regulate ADAM10 Synaptic Localization Through Aberrant Plasticity Phenomena. Mol. Neurobiol. 2019, 56, 7136–7143. [Google Scholar] [CrossRef] [Green Version]
- Tackenberg, C.; Grinschgl, S.; Trutzel, A.; Santuccione, A.C.; Frey, M.C.; Konietzko, U.; Grimm, J.; Brandt, R.; Nitsch, R.M. NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 2013, 4, e608. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franchini, L.; Carrano, N.; Di Luca, M.; Gardoni, F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 1538. https://doi.org/10.3390/ijms21041538
Franchini L, Carrano N, Di Luca M, Gardoni F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. International Journal of Molecular Sciences. 2020; 21(4):1538. https://doi.org/10.3390/ijms21041538
Chicago/Turabian StyleFranchini, Luca, Nicolò Carrano, Monica Di Luca, and Fabrizio Gardoni. 2020. "Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity" International Journal of Molecular Sciences 21, no. 4: 1538. https://doi.org/10.3390/ijms21041538
APA StyleFranchini, L., Carrano, N., Di Luca, M., & Gardoni, F. (2020). Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. International Journal of Molecular Sciences, 21(4), 1538. https://doi.org/10.3390/ijms21041538