Visualizing the Synaptic and Cellular Ultrastructure in Neurons Differentiated from Human Induced Neural Stem Cells—An Optimized Protocol

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
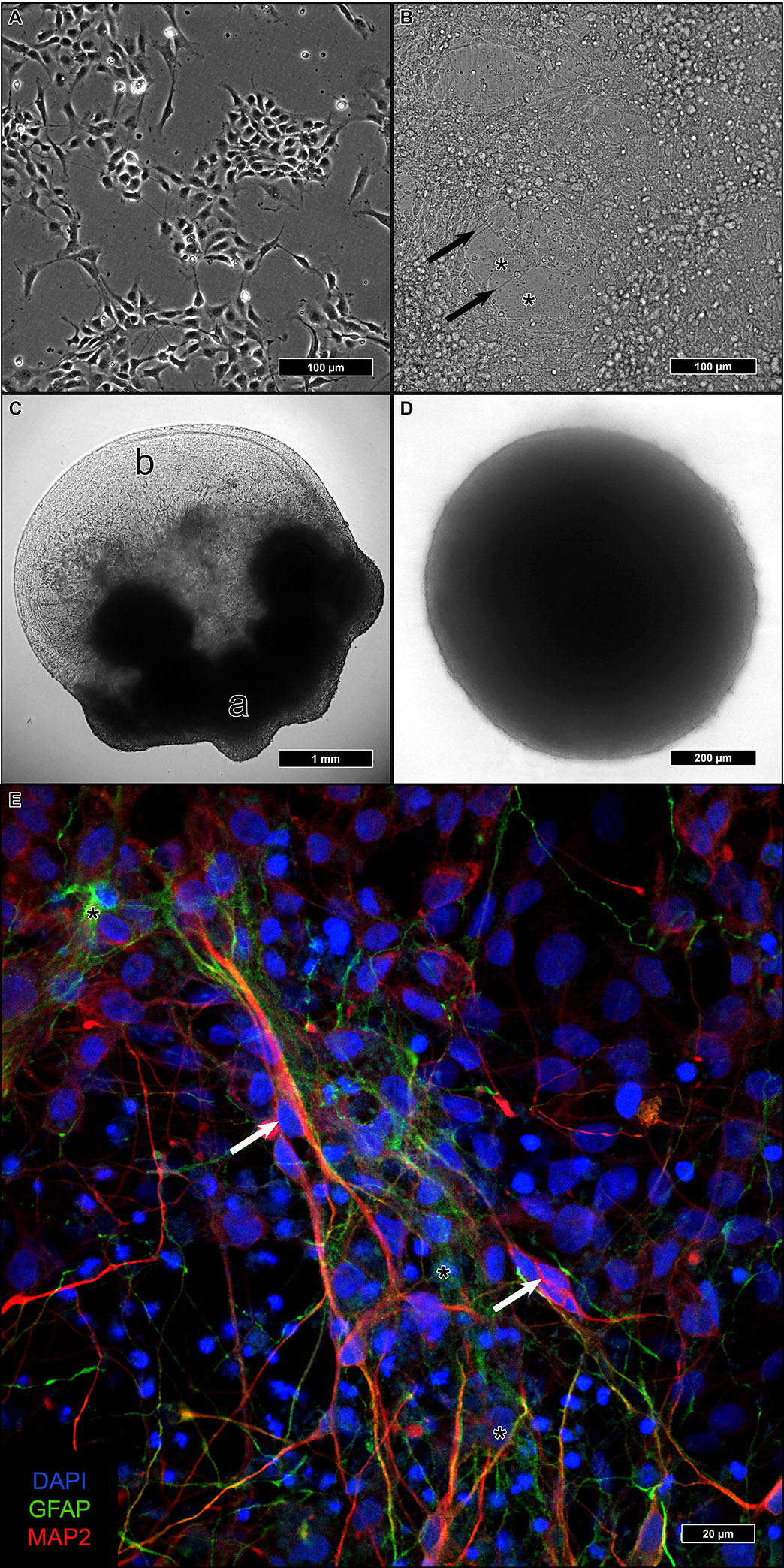
2.1. Differentiation of iNSC under Three Distinct Culture Conditions
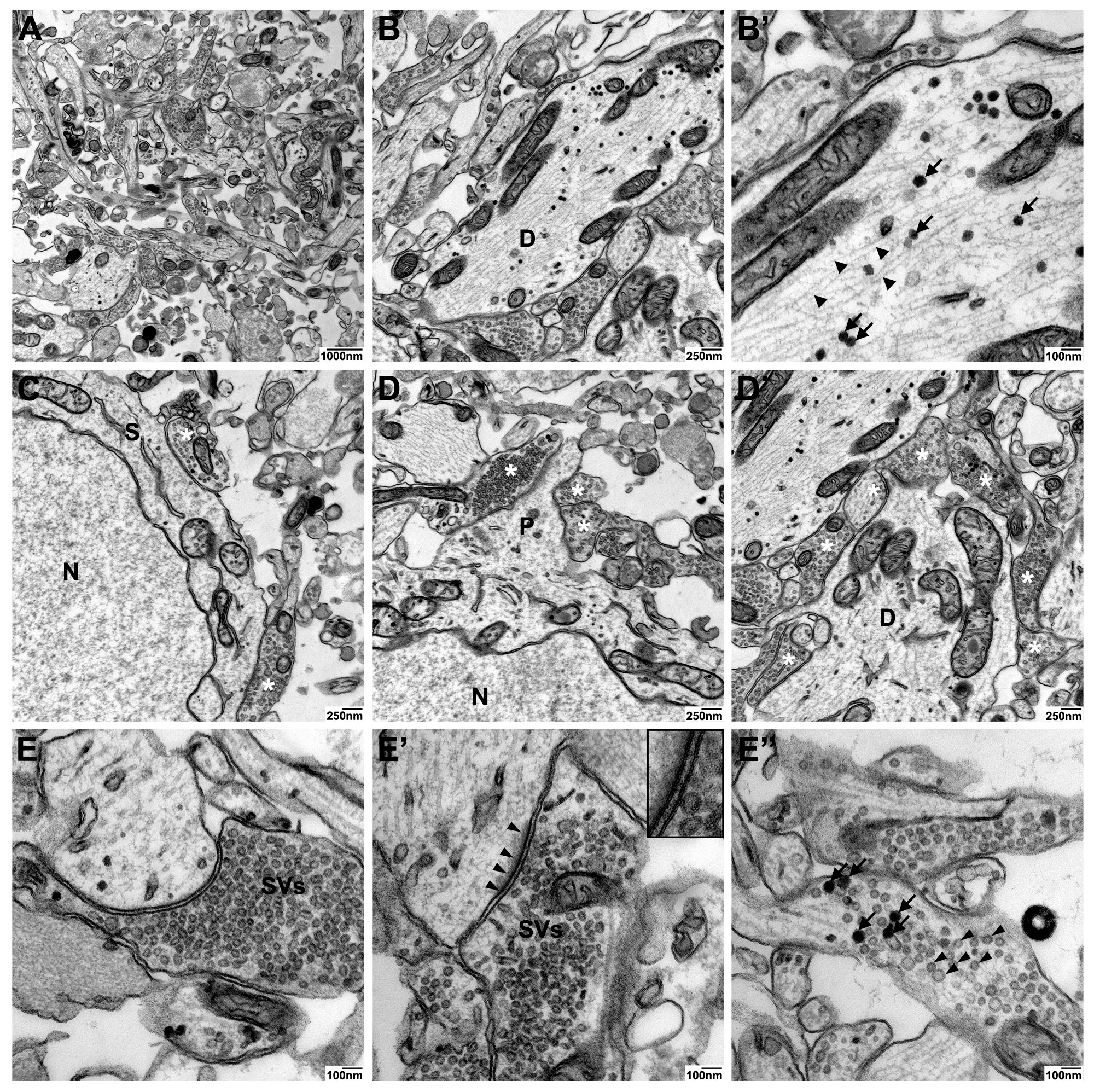
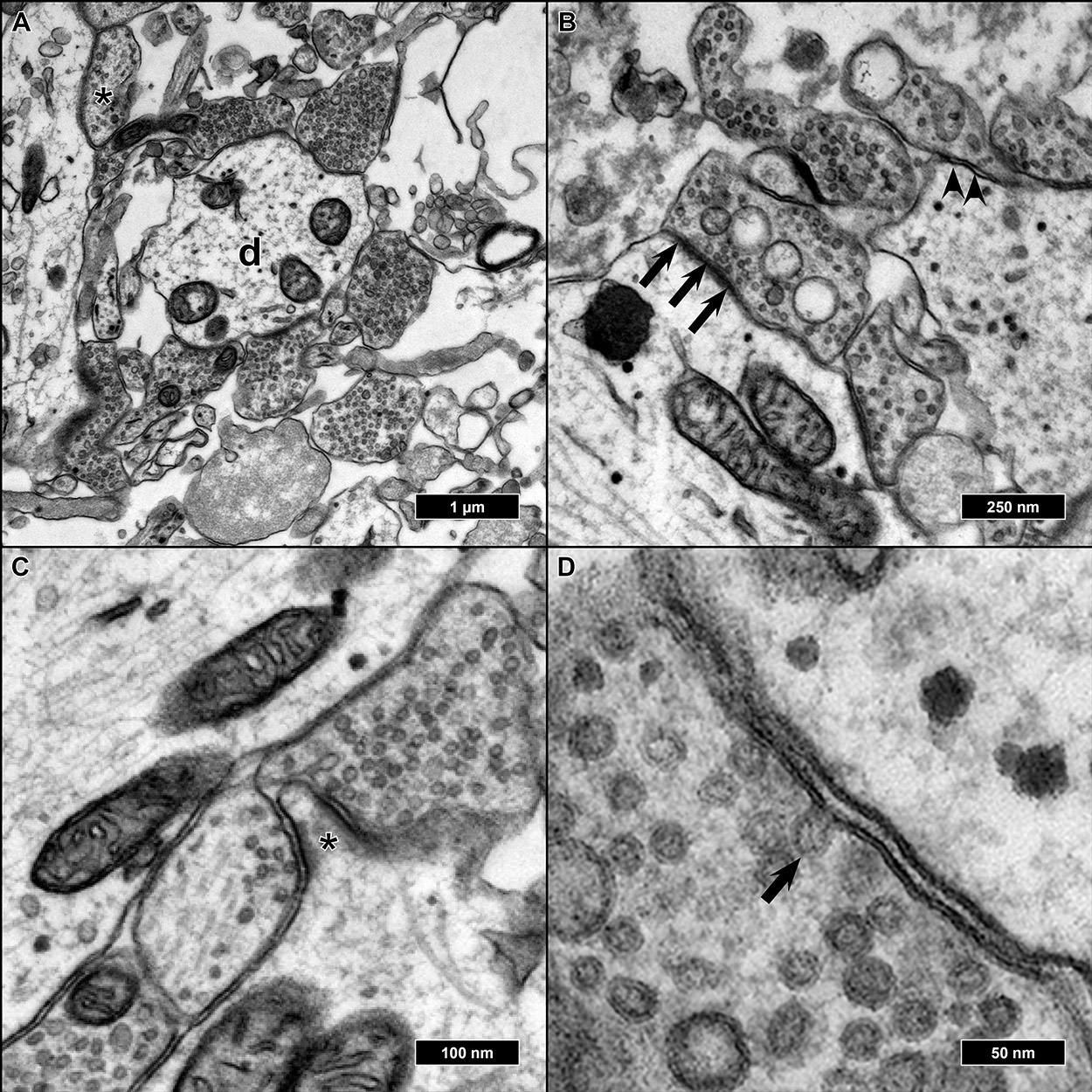
2.2. Ultrastructural Preservation under Three Different Culture Conditions
2.3. Comparison of Three EM Preparation Protocols
3. Discussion
4. Materials and Methods
4.1. Derivation and Proliferation of iNSC
4.2. Differentiation of iNSC
4.2.1. Adherent Differentiation on Glass Coverslips
4.2.2. Free Floating Differentiation of Aggregates Embedded in Matrigel
4.2.3. Free Floating Differentiation of Neurospheres
4.2.4. Replating and Immunofluorescence of Neurospheres
4.3. Specimen Preparation for Transmission Electron Microscopy
4.3.1. Standard Electron Microscopic Preparation (Standard Stain)
4.3.2. Electron Microscopic Preparation Using Ruthenium Red (Ruthenium Red Stain)
4.3.3. High Contrast Electron Microscopic Preparation (High Contrast En-Bloc Stain)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TEM | Transmission electron microscopy |
| GA | Glutaraldehyde |
| dSTORM | Direct stochastic optical reconstruction microscopy |
| EBNA-1 | Epstein–Barr virus nuclear antigen 1 |
| EBV | Epstein–Barr virus |
| GFAP | Glial fibrillary acidic protein |
| iNSC | Induced neural stem cells |
| MAP2 | Microtubule associated protein 2 |
| OriP | Origin of viral replication |
| PFA | Paraformaldehyde |
| PO | Propylene oxide |
| RT | Room temperature |
| UAR | Uranyl acetate replacement |
References
- Galbraith, C.G.; Galbraith, J.A. Super-resolution microscopy at a glance. J. Cell Sci. 2011, 124, 1607–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, A.J.; Genoud, C.; Pont, M.; van de Berg, W.D.; Frank, S.; Stahlberg, H.; Shahmoradian, S.H.; Al-Amoudi, A. Imaging of post-mortem human brain tissue using electron and X-ray microscopy. Curr. Opin. Struct. Biol. 2019, 58, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Lorenzo-Betancor, O.; Pastor, P.; Agúndez, J.a.G. Update on genetics of essential tremor. Acta Neurol. Scand. 2013, 128, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Efe, J.A.; Zhu, S.; Talantova, M.; Yuan, X.; Wang, S.; Lipton, S.A.; Zhang, K.; Ding, S. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc. Natl. Acad. Sci. 2011, 108, 7838–7843. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, F.; Stubblefield, E.A.; Blanchard, B.; Richards, T.L.; Larson, G.A.; He, Y.; Huang, Q.; Tan, A.-C.; Zhang, D.; et al. Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res. 2011. [Google Scholar] [CrossRef]
- Kim, J.-E.; O’Sullivan, M.L.; Sanchez, C.A.; Hwang, M.; Israel, M.A.; Brennand, K.; Deerinck, T.J.; Goldstein, L.S.B.; Gage, F.H.; Ellisman, M.H.; et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc. Natl. Acad. Sci. USA. 2011, 108, 3005–3010. [Google Scholar] [CrossRef] [Green Version]
- Capetian, P.; Azmitia, L.; Pauly, M.G.; Krajka, V.; Stengel, F.; Bernhardi, E.-M.; Klett, M.; Meier, B.; Seibler, P.; Stanslowsky, N.; et al. Plasmid-based generation of induced neural stem cells from adult human fibroblasts. Front. Cell. Neurosci. 2016, 10, 245. [Google Scholar] [CrossRef] [Green Version]
- Luft, J.H. Ruthenium red and violet. I. Chemistry, purification, methods of use for electron microscopy and mechanism of action. Anat. Rec. 1971, 171, 347–368. [Google Scholar] [CrossRef]
- Sobota, A.; Mrozińska, K.; Popov, V.I. Anionic domains on the cytoplasmic surface of the plasma membrane of Acanthamoeba castellanii and their relation to calcium-binding microregions. Acta Protozoologica 1997, 36, 187–196. [Google Scholar]
- Hadley, D.; Murphy, T.; Valladares, O.; Hannenhalli, S.; Ungar, L.; Kim, J.; Bućan, M. Patterns of sequence conservation in presynaptic neural genes. Genome Biol. 2006, 7, R105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.A.; Harrison, C.; Eaton, S.L.; Llavero Hurtado, M.; Graham, L.C.; Alkhammash, L.; Oladiran, O.A.; Gale, A.; Lamont, D.J.; Simpson, H.; et al. Cellular and Molecular Anatomy of the Human Neuromuscular Junction. Cell Rep. 2017, 21, 2348–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayés, À.; Collins, M.O.; Croning, M.D.R.; Lagemaat, L.N. van de; Choudhary, J.S.; Grant, S.G.N. Comparative Study of Human and Mouse Postsynaptic Proteomes Finds High Compositional Conservation and Abundance Differences for Key Synaptic Proteins. PLoS ONE 2012, 7, e46683. [Google Scholar] [CrossRef] [PubMed]
- Capetian, P.; Stanslowsky, N.; Bernhardi, E.; Grütz, K.; Domingo, A.; Brüggemann, N.; Naujock, M.; Seibler, P.; Klein, C.; Wegner, F. Altered glutamate response and calcium dynamics in iPSC-derived striatal neurons from XDP patients. Exp. Neurol. 2018, 308, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Bradford, A.B.; McNutt, P.M. Importance of being Nernst: Synaptic activity and functional relevance in stem cell-derived neurons. World J. Stem Cells 2015, 7, 899–921. [Google Scholar] [CrossRef]
- Wilson, E.S.; Newell-Litwa, K. Stem cell models of human synapse development and degeneration. Mol. Biol. Cell 2018, 29, 2913–2921. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; Pircs, K.; Barker, R.A.; Jakobsson, J.; Parmar, M. Direct Neuronal Reprogramming for Disease Modeling Studies Using Patient-Derived Neurons: What Have We Learned? Front. Neurosci. 2017, 11, 530. [Google Scholar] [CrossRef] [Green Version]
- Azmitia, L.; Capetian, P. Single-Step Plasmid Based Reprogramming of Human Dermal Fibroblasts to Induced Neural Stem Cells. Methods Mol. Biol. Clifton NJ 2018, 1842, 31–41. [Google Scholar]
- Molliver, M.E.; Kostović, I.; van der Loos, H. The development of synapses in cerebral cortex of the human fetus. Brain Res. 1973, 50, 403–407. [Google Scholar] [CrossRef]
- Petit, T.L.; LeBoutillier, J.C.; Alfano, D.P.; Becker, L.E. Synaptic development in the human fetus: A morphometric analysis of normal and Down’s syndrome neocortex. Exp. Neurol. 1984, 83, 13–23. [Google Scholar] [CrossRef]
- Yuste, R.; Bonhoeffer, T. Genesis of dendritic spines: Insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 2004, 5, 24–34. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Yang, S.M.; Berger, D.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Spacek, J.; Harris, K.M. Three-Dimensional Organization of Smooth Endoplasmic Reticulum in Hippocampal CA1 Dendrites and Dendritic Spines of the Immature and Mature Rat. J. Neurosci. 1997, 17, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.B.; Parmar, M. Strengths and limitations of the neurosphere culture system. Mol. Neurobiol. 2006, 34, 153–161. [Google Scholar] [CrossRef]
- Pauly, M.G.; Krajka, V.; Stengel, F.; Seibler, P.; Klein, C.; Capetian, P. Adherent vs. Free-Floating Neural Induction by Dual SMAD Inhibition for Neurosphere Cultures Derived from Human Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Deerinck, T.J.; Bushong, E.A.; Thor, A.; Ellisman, M.H.; Deerinck, T.J.; Bushong, E.; Ellisman, M.; Deerinck, T.; Thor, A.; Thor, C.A. NCMIR Methods for 3D EM: A New Protocol for Preparation of Biological Specimens for Serial Block Face Scanning Electron Microscopy; Center for Research in Biological Systems and the National Center for Microscopy and Imaging Research, University of California: San Diego, CA, USA, 2010. [Google Scholar]
- Hua, Y.; Laserstein, P.; Helmstaedter, M. Large-volume en-bloc staining for electron microscopy-based connectomics. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Glauert, A.M.; Lucy, J.A. Electron microscopy of lipids: Effects of pH and fixatives on the appearance of a macromolecular assembly of lipid micelles in negatively stained preparations. J. Microsc. 1969, 89, 1–18. [Google Scholar] [CrossRef]
- Neiss, W.F. Electron staining of the cell surface coat by osmium-low ferrocyanide. Histochemistry 1984, 80, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Willingham, M.C.; Rutherford, A.V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1984, 32, 455–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakakoshi, M.; Nishioka, H.; Katayama, E. New versatile staining reagents for biological transmission electron microscopy that substitute for uranyl acetate. J. Electron Microsc. (Tokyo) 2011, 60, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Kopriwa, B.M. Block-staining tissues with potassium ferrocyanide-reduced osmium tetroxide and lead aspartate for electron microscopic radioautography. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1984, 32, 552–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1979, 27, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capetian, P.; Müller, L.; Volkmann, J.; Heckmann, M.; Ergün, S.; Wagner, N. Visualizing the Synaptic and Cellular Ultrastructure in Neurons Differentiated from Human Induced Neural Stem Cells—An Optimized Protocol. Int. J. Mol. Sci. 2020, 21, 1708. https://doi.org/10.3390/ijms21051708
Capetian P, Müller L, Volkmann J, Heckmann M, Ergün S, Wagner N. Visualizing the Synaptic and Cellular Ultrastructure in Neurons Differentiated from Human Induced Neural Stem Cells—An Optimized Protocol. International Journal of Molecular Sciences. 2020; 21(5):1708. https://doi.org/10.3390/ijms21051708
Chicago/Turabian StyleCapetian, Philipp, Lorenz Müller, Jens Volkmann, Manfred Heckmann, Süleyman Ergün, and Nicole Wagner. 2020. "Visualizing the Synaptic and Cellular Ultrastructure in Neurons Differentiated from Human Induced Neural Stem Cells—An Optimized Protocol" International Journal of Molecular Sciences 21, no. 5: 1708. https://doi.org/10.3390/ijms21051708
APA StyleCapetian, P., Müller, L., Volkmann, J., Heckmann, M., Ergün, S., & Wagner, N. (2020). Visualizing the Synaptic and Cellular Ultrastructure in Neurons Differentiated from Human Induced Neural Stem Cells—An Optimized Protocol. International Journal of Molecular Sciences, 21(5), 1708. https://doi.org/10.3390/ijms21051708