Roles of Splicing Factors in Hormone-Related Cancer Progression


Abstract
:1. Introduction
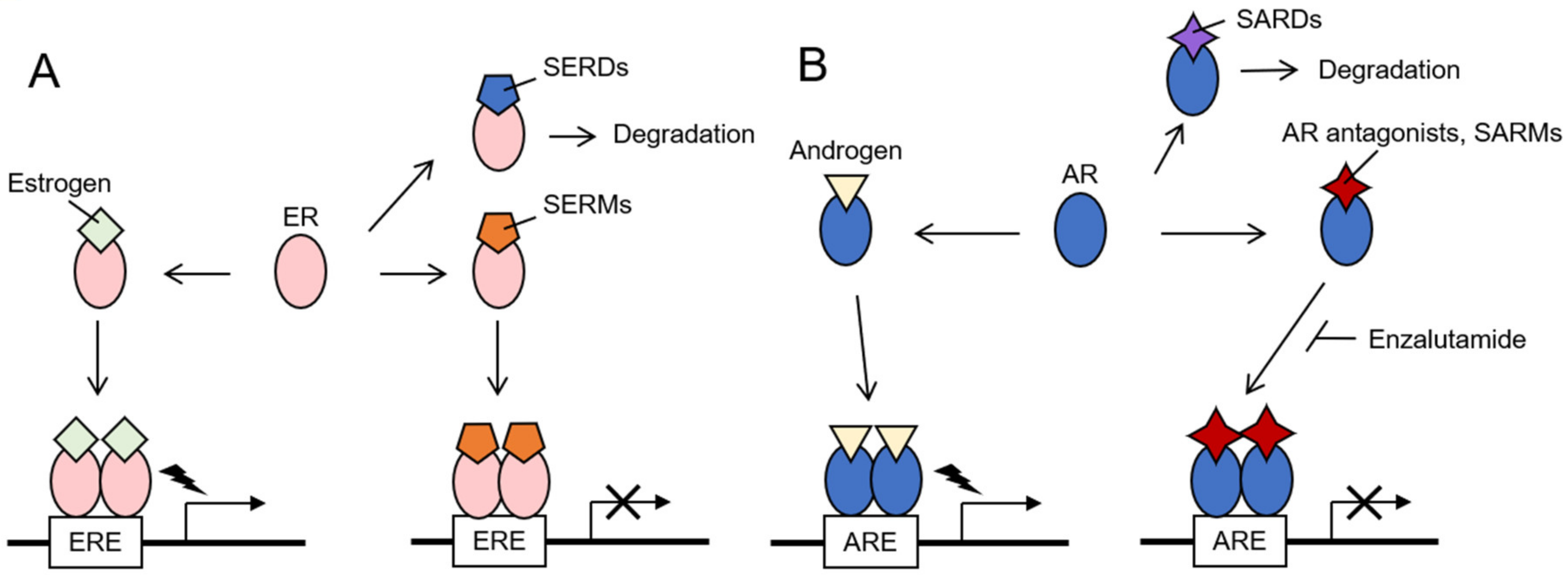
2. Hormone-Related Cancers
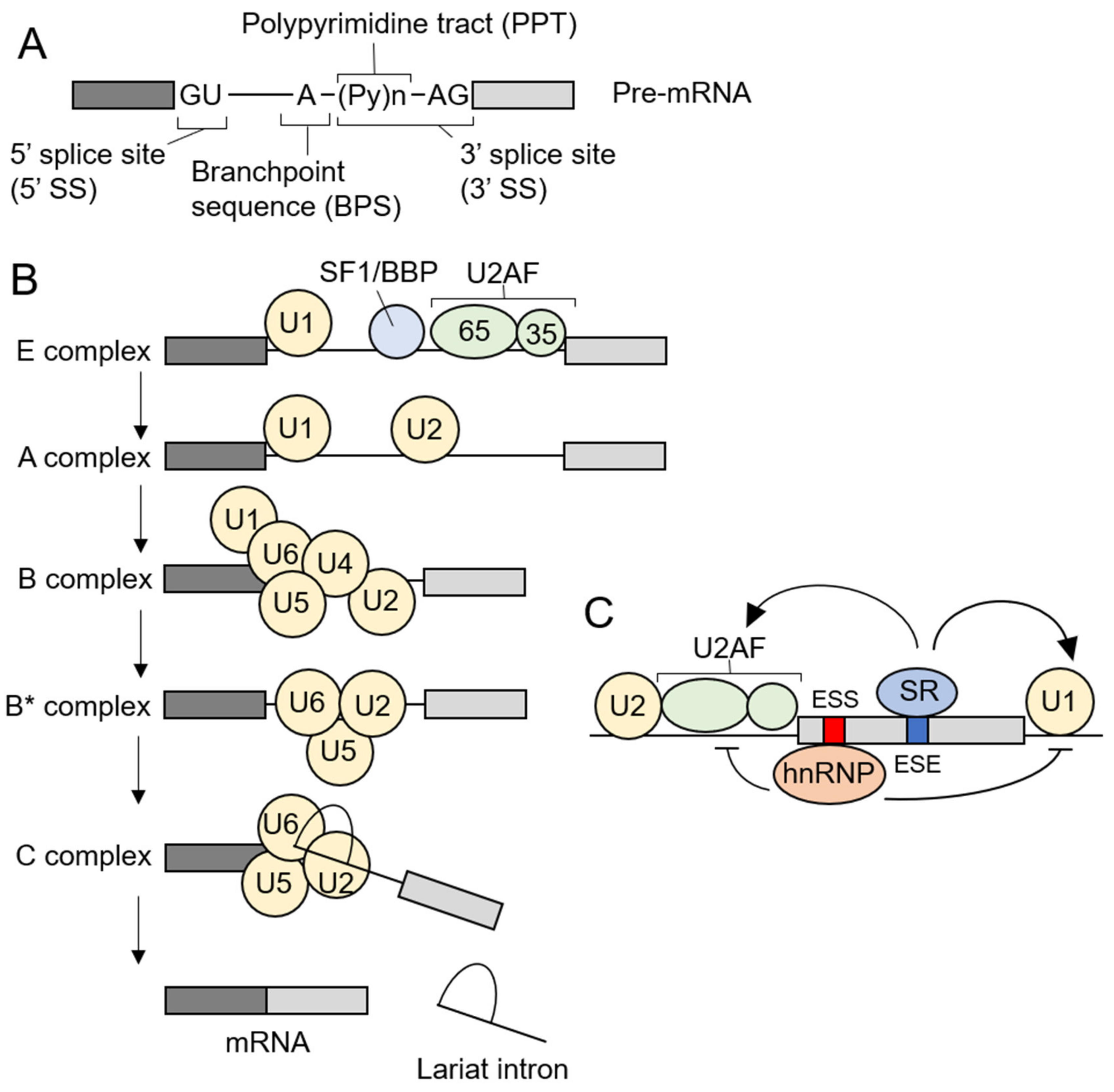
3. Pre-mRNA Splicing and Spliceosome Assembly
4. Alteration of Splicing Factors in Cancer
4.1. Somatic Mutations of Splicing Factors in Cancer
4.2. Deregulated Gene Expression of Splicing Factors in Cancer
5. The Roles of Splicing Factors and Splicing Regulatory Proteins in Breast and Prostate Cancers
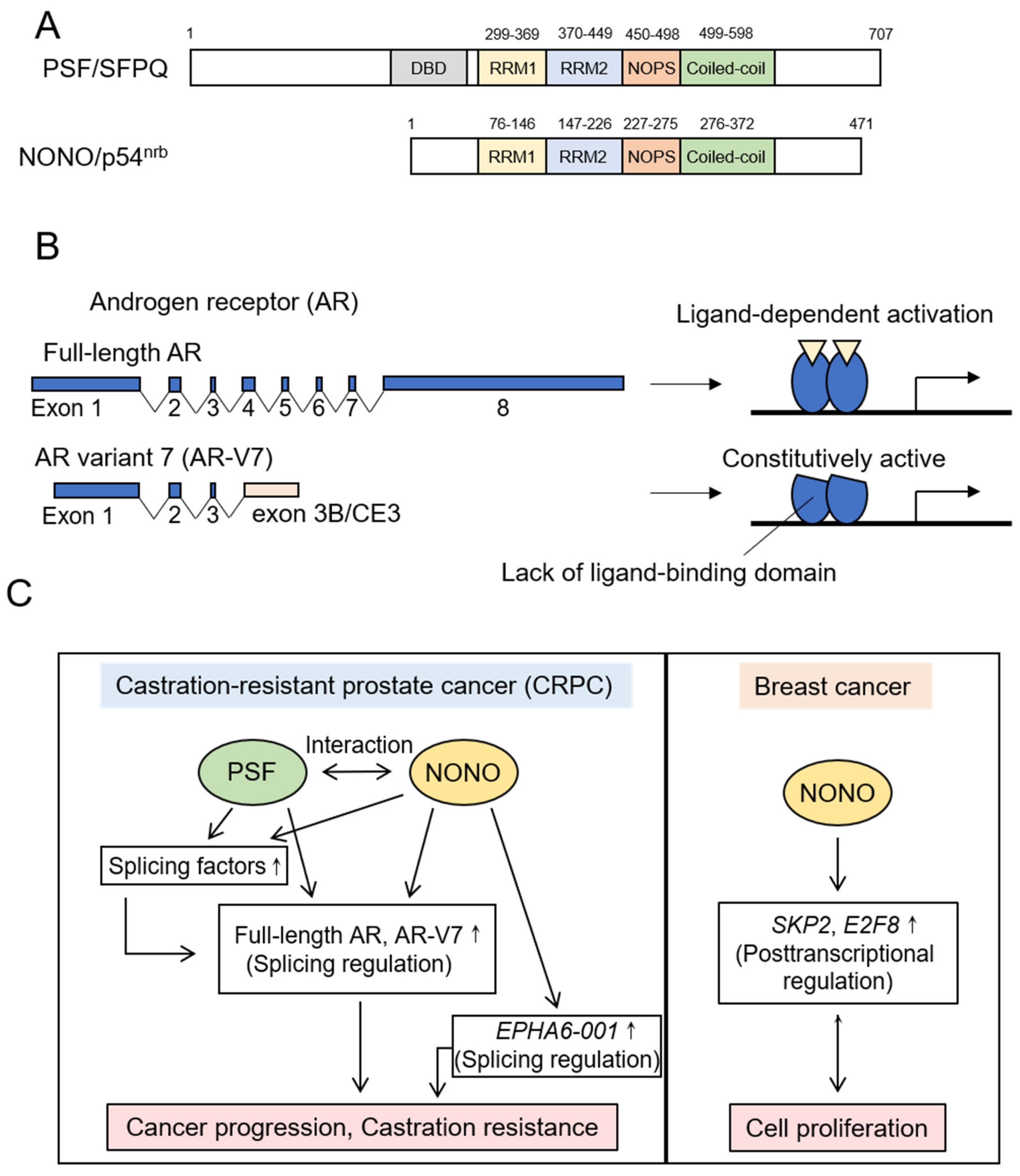
5.1. DBHS Family Proteins
5.1.1. PSF
5.1.2. NONO
5.2. SR Proteins
5.3. hnRNPs
5.4. Other Splicing Factors and Splicing Regulatory Proteins in Breast and Prostate Cancers
5.4.1. SF3b Complex
5.4.2. Src Associated in Mitosis of 68 kDa (Sam68)
5.4.3. SR Protein Kinases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [Green Version]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and transcription touch base: Co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Anczuków, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.A.; Yu, L.; Smith, P.G.; Buonamici, S. Targeting splicing abnormalities in cancer. Curr. Opin. Genet. Dev. 2018, 48, 67–74. [Google Scholar] [CrossRef]
- Song, X.; Zeng, Z.; Wei, H.; Wang, Z. Alternative splicing in cancers: From aberrant regulation to new therapeutics. Semin. Cell Dev. Biol. 2018, 75, 13–22. [Google Scholar] [CrossRef]
- Takayama, K.I. Splicing Factors Have an Essential Role in Prostate Cancer Progression and Androgen Receptor Signaling. Biomolecules 2019, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hilton, H.N.; Clarke, C.L.; Graham, J.D. Estrogen and progesterone signalling in the normal breast and its implications for cancer development. Mol. Cell. Endocrinol. 2018, 466, 2–14. [Google Scholar] [CrossRef]
- Brisken, C. Progesterone signalling in breast cancer: A neglected hormone coming into the limelight. Nat. Rev. Cancer 2013, 13, 385–396. [Google Scholar] [CrossRef]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef] [Green Version]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2013, 20, R49–R64. [Google Scholar] [CrossRef] [Green Version]
- Chmelar, R.; Buchanan, G.; Need, E.F.; Tilley, W.; Greenberg, N.M. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer 2007, 120, 719–733. [Google Scholar] [CrossRef]
- Renoir, J.M.; Marsaud, V.; Lazennec, G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem. Pharmacol. 2013, 85, 449–465. [Google Scholar] [CrossRef] [Green Version]
- Leonhardt, S.A.; Boonyaratanakornkit, V.; Edwards, D.P. Progesterone receptor transcription and non-transcription signaling mechanisms. Steroids 2003, 68, 761–770. [Google Scholar] [CrossRef]
- Gao, X.; Nawaz, Z. Progesterone receptors-animal models and cell signaling in breast cancer: Role of steroid receptor coactivators and corepressors of progesterone receptors in breast cancer. Breast Cancer Res. 2002, 4, 182–186. [Google Scholar] [CrossRef]
- Reinbolt, R.E.; Mangini, N.; Hill, J.L.; Levine, L.B.; Dempsey, J.L.; Singaravelu, J.; Koehler, K.A.; Talley, A.; Lustberg, M.B. Endocrine therapy in breast cancer: The neoadjuvant, adjuvant, and metastatic approach. Semin. Oncol. Nurs. 2015, 31, 146–155. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Damber, J.E. Endocrine therapy for prostate cancer. Acta. Oncol. 2005, 44, 605–609. [Google Scholar] [CrossRef]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- Solomon, Z.J.; Mirabal, J.R.; Mazur, D.J.; Kohn, T.P.; Lipshultz, L.I.; Pastuszak, A.W. Selective Androgen Receptor Modulators: Current Knowledge and Clinical Applications. Sex. Med. Rev. 2019, 7, 84–94. [Google Scholar] [CrossRef]
- Narayanan, R.; Ponnusamy, S.; Miller, D.D. Destroying the androgen receptor (AR)-potential strategy to treat advanced prostate cancer. Oncoscience 2017, 4, 175–177. [Google Scholar]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [Green Version]
- Verma, B.; Akinyi, M.V.; Norppa, A.J.; Frilander, M.J. Minor spliceosome and disease. Semin. Cell Dev. Biol. 2018, 79, 103–112. [Google Scholar] [CrossRef]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Turunen, J.J.; Niemelä, E.H.; Verma, B.; Frilander, M.J. The significant other: Splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA 2013, 4, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Zheng, X.; Luecke, S.; Green, M.R. The U2AF35-related protein Urp contacts the 3’ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev. 2010, 24, 2389–2394. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Ogawa, S. Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA 2014, 5, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Visconte, V.; Makishima, H.; Jankowska, A.; Szpurka, H.; Traina, F.; Jerez, A.; O’Keefe, C.; Rogers, H.J.; Sekeres, M.A.; Maciejewski, J.P.; et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 2012, 26, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Rossi, D.; Bruscaggin, A.; Spina, V.; Rasi, S.; Khiabanian, H.; Messina, M.; Fangazio, M.; Vaisitti, T.; Monti, S.; Chiaretti, S.; et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: Association with progression and fludarabine-refractoriness. Blood 2011, 118, 6904–6908. [Google Scholar] [CrossRef] [Green Version]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef] [Green Version]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef]
- Przychodzen, B.; Jerez, A.; Guinta, K.; Sekeres, M.A.; Padgett, R.; Maciejewski, J.P.; Makishima, H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 2013, 122, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.N.; Choi, P.S.; de Waal, L.; Sharifnia, T.; Imielinski, M.; Saksena, G.; Pedamallu, C.S.; Sivachenko, A.; Rosenberg, M.; Chmielecki, J.; et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE 2014, 9, e87361. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef]
- Okeyo-Owuor, T.; White, B.S.; Chatrikhi, R.; Mohan, D.R.; Kim, S.; Griffith, M.; Ding, L.; Ketkar-Kulkarni, S.; Hundal, J.; Laird, K.M.; et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 2015, 29, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Shirai, C.L.; White, B.S.; Tripathi, M.; Tapia, R.; Ley, J.N.; Ndonwi, M.; Kim, S.; Shao, J.; Carver, A.; Saez, B.; et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun. 2017, 8, 14060. [Google Scholar] [CrossRef]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Haferlach, T.; et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhan, Z.; Naren, D.; Li, J.; Yan, T.; Gong, Y. Prognostic value of SRSF2 mutations in patients with de novo myelodysplastic syndromes: A meta-analysis. PLoS ONE 2017, 12, e0185053. [Google Scholar] [CrossRef] [Green Version]
- Arbab Jafari, P.; Ayatollahi, H.; Sadeghi, R.; Sheikhi, M.; Asghari, A. Prognostic significance of SRSF2 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: A meta-analysis. Hematology 2018, 23, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; De Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef]
- Ke, H.; Zhao, L.; Zhang, H.; Feng, X.; Xu, H.; Hao, J.; Wang, S.; Yang, Q.; Zou, L.; Su, X.; et al. Loss of TDP43 inhibits progression of triple-negative breast cancer in coordination with SRSF3. Proc. Natl. Acad. Sci. USA 2018, 115, E3426–E3435. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Langiewicz, M.; Jumaa, H.; Webster, N.J. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015, 61, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Bond, C.S.; Fox, A.H. The DBHS proteins SFPQ, NONO and PSPC1: A multipurpose molecular scaffold. Nucleic Acids Res. 2016, 44, 3989–4004. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Sadowska, A.; Bekere, I.; Ho, D.; Gully, B.S.; Lu, Y.; Iyer, K.S.; Trewhella, J.; Fox, A.H.; Bond, C.S. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Res. 2015, 43, 3826–3840. [Google Scholar] [CrossRef]
- Takayama, K.I.; Suzuki, T.; Fujimura, T.; Yamada, Y.; Takahashi, S.; Homma, Y.; Suzuki, Y.; Inoue, S. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. Proc. Natl. Acad. Sci. USA 2017, 114, 10461–10466. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Armstrong, A.J.; Dehm, S.M.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Yu, B.; Liu, Y. Androgen receptor splicing variant 7: Beyond being a constitutively active variant. Life Sci. 2019, 234, 116768. [Google Scholar] [CrossRef]
- Sprenger, C.C.; Plymate, S.R. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm. Cancer 2014, 5, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Seisen, T.; Rouprêt, M.; Gomez, F.; Malouf, G.G.; Shariat, S.F.; Peyronnet, B.; Spano, J.P.; Cancel-Tassin, G.; Cussenot, O. A comprehensive review of genomic landscape, biomarkers and treatment sequencing in castration-resistant prostate cancer. Cancer Treat. Rev. 2016, 48, 25–33. [Google Scholar] [CrossRef]
- Bastos, D.A.; Antonarakis, E.S. CTC-derived AR-V7 detection as a prognostic and predictive biomarker in advanced prostate cancer. Expert Rev. Mol. Diagn. 2018, 18, 155–163. [Google Scholar] [CrossRef]
- Okegawa, T.; Ninomiya, N.; Masuda, K.; Nakamura, Y.; Tambo, M.; Nutahara, K. AR-V7 in circulating tumor cells cluster as a predictive biomarker of abiraterone acetate and enzalutamide treatment in castration-resistant prostate cancer patients. Prostate 2018, 78, 576–582. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarese, C.; Santoni, M.; Brunelli, M.; Buti, S.; Modena, A.; Nabissi, M.; Artibani, W.; Martignoni, G.; Montironi, R.; Tortora, G.; et al. AR-V7 and prostate cancer: The watershed for treatment selection? Cancer Treat. Rev. 2016, 43, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Iino, K.; Mitobe, Y.; Ikeda, K.; Takayama, K.I.; Suzuki, T.; Kawabata, H.; Suzuki, Y.; Horie-Inoue, K.; Inoue, S. RNA-binding protein NONO promotes breast cancer proliferation by post-transcriptional regulation of SKP2 and E2F8. Cancer Sci. 2020, 111, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Osawa, T.; Sasaki, Y.; Yamamoto, S.; Anai, M.; Izumi, K.; Matsumura, Y.; Sakai, J.; Aburatani, H.; Mizokami, A.; et al. Overexpression of p54. Oncotarget 2018, 9, 10510–10524. [Google Scholar]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Gautrey, H.L.; Tyson-Capper, A.J. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PLoS ONE 2012, 7, e51497. [Google Scholar] [CrossRef]
- Anczuków, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [Green Version]
- Olshavsky, N.A.; Comstock, C.E.; Schiewer, M.J.; Augello, M.A.; Hyslop, T.; Sette, C.; Zhang, J.; Parysek, L.M.; Knudsen, K.E. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010, 70, 3975–3984. [Google Scholar] [CrossRef] [Green Version]
- Comstock, C.E.; Augello, M.A.; Benito, R.P.; Karch, J.; Tran, T.H.; Utama, F.E.; Tindall, E.A.; Wang, Y.; Burd, C.J.; Groh, E.M.; et al. Cyclin D1 splice variants: Polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin. Cancer Res. 2009, 15, 5338–5349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.J.; Nishi, K.; Isono, T.; Okuyama, Y.; Tambe, Y.; Okada, Y.; Inoue, H. Cyclin D1b variant promotes cell invasiveness independent of binding to CDK4 in human bladder cancer cells. Mol. Carcinog. 2009, 48, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Millar, E.K.; Dean, J.L.; McNeil, C.M.; O’Toole, S.A.; Henshall, S.M.; Tran, T.; Lin, J.; Quong, A.; Comstock, C.E.; Witkiewicz, A.; et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene 2009, 28, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, G.; Difeo, A.; Reeves, H.L.; Schaid, D.J.; Hirshfeld, J.; Hod, E.; Katz, A.; Isaacs, W.B.; Hebbring, S.; Komiya, A.; et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res. 2005, 65, 1213–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, G.; DiFeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Brugiolo, M.; Akerman, M.; Das, S.; Urbanski, L.; Geier, A.; Kesarwani, A.K.; Fan, M.; Leclair, N.; Lin, K.T.; et al. Differential Functions of Splicing Factors in Mammary Transformation and Breast Cancer Metastasis. Cell Rep. 2019, 29, 2672–2688. [Google Scholar] [CrossRef]
- Gabriel, M.; Delforge, Y.; Deward, A.; Habraken, Y.; Hennuy, B.; Piette, J.; Klinck, R.; Chabot, B.; Colige, A.; Lambert, C. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer 2015, 15, 227. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Zhang, F.; Xu, S.; Cui, X.; Hussain, A.; Fazli, L.; Gleave, M.; Dong, X.; Qi, J. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4584–E4593. [Google Scholar] [CrossRef] [Green Version]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Tummala, R.; Lou, W.; Gao, A.C.; Nadiminty, N. Quercetin Targets hnRNPA1 to Overcome Enzalutamide Resistance in Prostate Cancer Cells. Mol. Cancer Ther. 2017, 16, 2770–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, N.; Huang, J.; Ho, T.T.; Zhu, Z.; Qiu, Z.; Zhou, X.; Bai, C.; Wu, F.; Xu, M.; et al. Regulation of androgen receptor splice variant AR3 by PCGEM1. Oncotarget 2016, 7, 15481–15491. [Google Scholar] [CrossRef] [PubMed]
- Fei, T.; Chen, Y.; Xiao, T.; Li, W.; Cato, L.; Zhang, P.; Cotter, M.B.; Bowden, M.; Lis, R.T.; Zhao, S.G.; et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, E5207–E5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, Q.; He, J.; Zhong, L.; Shu, F.; Xing, R.; Lv, D.; Lei, B.; Wan, B.; Yang, Y.; et al. HnRNP-L promotes prostate cancer progression by enhancing cell cycling and inhibiting apoptosis. Oncotarget 2017, 8, 19342–19353. [Google Scholar] [CrossRef] [PubMed]
- Tyson-Capper, A.; Gautrey, H. Regulation of Mcl-1 alternative splicing by hnRNP F, H1 and K in breast cancer cells. RNA Biol. 2018, 15, 1448–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Gao, X.D.; Lee, J.H.; Huang, H.; Tan, H.; Ahn, J.; Reinke, L.M.; Peter, M.E.; Feng, Y.; Gius, D.; et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014, 28, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Tian, Y.; Yuan, X.; Wu, H.; Liu, Q.; Pestell, R.G.; Wu, K. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco. Targets Ther. 2015, 8, 3783–3792. [Google Scholar]
- Zhang, F.L.; Cao, J.L.; Xie, H.Y.; Sun, R.; Yang, L.F.; Shao, Z.M.; Li, D.Q. Cancer-Associated MORC2-Mutant M276I Regulates an hnRNPM-Mediated CD44 Splicing Switch to Promote Invasion and Metastasis in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 5780–5792. [Google Scholar] [CrossRef] [Green Version]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.; Sapino, A.; Salomon, A.V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Gökmen-Polar, Y.; Neelamraju, Y.; Goswami, C.P.; Gu, X.; Nallamothu, G.; Janga, S.C.; Badve, S. Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen receptor-positive breast cancer. Mod. Pathol. 2015, 28, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.Z.; Xue, M.Z.; Shen, H.J.; Li, X.G.; Ma, D.; Gong, Y.; Liu, Y.R.; Qiao, F.; Xie, H.Y.; Lian, B.; et al. PHF5A Epigenetically Inhibits Apoptosis to Promote Breast Cancer Progression. Cancer Res. 2018, 78, 3190–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, N.; Nimura, K.; Saga, K.; Ishibashi, A.; Kitamura, K.; Nagano, H.; Yoshikawa, Y.; Ishida, K.; Nonomura, N.; Arisawa, M.; et al. SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression. Cancer Res. 2019, 79, 5204–5217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Gómez-Gómez, E.; León-González, A.J.; Sáez-Martínez, P.; Alors-Pérez, E.; Fuentes-Fayos, A.C.; Martínez-López, A.; Sánchez-Sánchez, R.; González-Serrano, T.; et al. Spliceosome component SF3B1 as novel prognostic biomarker and therapeutic target for prostate cancer. Transl. Res. 2019, 212, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Bielli, P.; Busà, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr. Relat. Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huot, M.E.; Vogel, G.; Richard, S. Identification of a Sam68 ribonucleoprotein complex regulated by epidermal growth factor. J. Biol. Chem. 2009, 284, 31903–31913. [Google Scholar] [CrossRef] [Green Version]
- Cappellari, M.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Saarikettu, J.; Silvennoinen, O.; Sette, C. The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 2014, 33, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Cappellari, M.; Busà, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.E.; Sette, C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010, 70, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Stockley, J.; Markert, E.; Zhou, Y.; Robson, C.N.; Elliott, D.J.; Lindberg, J.; Leung, H.Y.; Rajan, P. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci. Rep. 2015, 5, 13426. [Google Scholar] [CrossRef]
- Bielli, P.; Busà, R.; Di Stasi, S.M.; Munoz, M.J.; Botti, F.; Kornblihtt, A.R.; Sette, C. The transcription factor FBI-1 inhibits SAM68-mediated BCL-X alternative splicing and apoptosis. EMBO Rep. 2014, 15, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Babic, I.; Jakymiw, A.; Fujita, D.J. The RNA binding protein Sam68 is acetylated in tumor cell lines, and its acetylation correlates with enhanced RNA binding activity. Oncogene 2004, 23, 3781–3789. [Google Scholar] [CrossRef] [Green Version]
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, E3374–E3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, K.; Urano, T.; Horie-Inoue, K.; Hayashi, S.; Sakai, R.; Ouchi, Y.; Inoue, S. Association of estrogen receptor alpha and histone deacetylase 6 causes rapid deacetylation of tubulin in breast cancer cells. Cancer Res. 2009, 69, 2935–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, G.M.; Carrigan, P.E.; Beck, A.M.; Miller, L.J. Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res. 2006, 66, 3819–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, G.M.; Carrigan, P.E.; Miller, L.J. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007, 67, 2072–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, N.; Potts, J.; Simpkin, A.J.; Koupparis, A.; Harper, S.J.; Oxley, J.; Oltean, S. Serine-arginine protein kinase 1 (SRPK1), a determinant of angiogenesis, is upregulated in prostate cancer and correlates with disease stage and invasion. J. Clin. Pathol. 2016, 69, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roosmalen, W.; Le Dévédec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; de Graauw, M.; et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J. Clin. Invest. 2015, 125, 1648–1664. [Google Scholar] [CrossRef] [Green Version]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.; Oltean, S. Modulation of VEGF-A Alternative Splicing as a Novel Treatment in Chronic Kidney Disease. Genes (Basel) 2018, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.C.; Lin, C.Y.; Tarn, W.Y.; Li, F.Y. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. RNA 2014, 20, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, Y.J.; Liu, Z.Z.; Wan, S.; Cai, Z.D.; Xie, J.J.; Song, S.D.; Wan, Y.P.; Hua, W.; Zhong, W.; Wu, C.L. Enhanced expression of SRPK2 contributes to aggressive progression and metastasis in prostate cancer. Biomed. Pharmacother. 2018, 102, 531–538. [Google Scholar] [CrossRef]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Czubaty, A.; Piekiełko-Witkowska, A. Protein kinases that phosphorylate splicing factors: Roles in cancer development, progression and possible therapeutic options. Int. J. Biochem. Cell Biol. 2017, 91, 102–115. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Splicing Factor | Target Genes | Effects on Cancer Progression | References |
|---|---|---|---|---|
| Breast cancer | NONO/p54nrb | SKP2 and E2F8 | Upregulating cell proliferation | [74] |
| SRSF1 | Ron | Activating cell migration | [76] | |
| BIM, BIN1 | Upregulating cell proliferation Inhibiting apoptosis | [78] | ||
| Mnk2 | [78] | |||
| Mcl-1 | [77] | |||
| SRSF3 | PAR3 | Activating cell migration and invasion | [60] | |
| NUMB | Upregulate cell proliferation | [60] | ||
| HER2 | Possibly upregulating cell proliferation | [86] | ||
| SRSF4 | Cisplatin-induced splicing events including hnRNPDL and AMZ2 | Cisplatin-induced cell death | [88] | |
| SRSF4, 6 | Transformation-associated genes | Activating cell migration and invasion | [87] | |
| hnRNPF, H1, K | Mcl-1 | Inhibiting apoptosis | [95] | |
| hnRNPH1 | HER2 | Possibly upregulating cell proliferation | [86] | |
| hnRNPM | CD44 | Upregulating EMT and tumor metastasis | [96,98] | |
| SF3B3 | Associating with poor relapse-free and overall survival in ER-positive breast cancer patients | [100] | ||
| PHF5A/SF3B7 | Multiple genes involved in apoptosis including FASTK | Inhibiting apoptosis Associating with poor disease-free survival in breast cancer patients | [101] | |
| Sam68 | CD44 | Upregulating tumor metastasis | [105] | |
| SRPK1 | Mcl-1 and IR | Inhibiting apoptosis | [119] | |
| CLKs | p53 | Regulating cell proliferation | [121] | |
| CLK2 | EMT-related genes including ENAH | Suppressing cell migration and invasion Upregulating cell proliferation | [123] | |
| Prostate Cancer | PSF/SFPQ | AR, spliceosome genes | Upregulating cell proliferation Promoting CRPC progression | [64] |
| NONO/p54nrb | AR, spliceosome genes, EPHA6 | Upregulating cell proliferation Promoting CRPC progression | [64,75] | |
| SRSF1 | AR | [79] | ||
| CCND1 | [80] | |||
| SRSF5 | KLF6 | Upregulating cell proliferation, migration, and invasion | [84,85] | |
| hnRNPA1 | AR | [90,91,92] | ||
| hnRNPF | AR | Possibly upregulating cell proliferation and inhibiting apoptosis | [89] | |
| hnRNPL | Various genes including AR and MYH10 | Upregulating cell proliferation Inhibiting apoptosis | [93,94] | |
| SF3B1 | AR, Ghrelin | Upregulating cell proliferation, survival, migration | [103] | |
| SF3B2 | AR | Promoting CRPC progression | [102] | |
| Sam68 | CD44 | [106] | ||
| CCND1 | [107] | |||
| AR | [108] | |||
| BCL-X | Promoting apoptosis | [109] | ||
| SRPK1 | VEGF-A | Upregulating tumor growth via angiogenesis | [117] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeiwa, T.; Mitobe, Y.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Roles of Splicing Factors in Hormone-Related Cancer Progression. Int. J. Mol. Sci. 2020, 21, 1551. https://doi.org/10.3390/ijms21051551
Takeiwa T, Mitobe Y, Ikeda K, Horie-Inoue K, Inoue S. Roles of Splicing Factors in Hormone-Related Cancer Progression. International Journal of Molecular Sciences. 2020; 21(5):1551. https://doi.org/10.3390/ijms21051551
Chicago/Turabian StyleTakeiwa, Toshihiko, Yuichi Mitobe, Kazuhiro Ikeda, Kuniko Horie-Inoue, and Satoshi Inoue. 2020. "Roles of Splicing Factors in Hormone-Related Cancer Progression" International Journal of Molecular Sciences 21, no. 5: 1551. https://doi.org/10.3390/ijms21051551
APA StyleTakeiwa, T., Mitobe, Y., Ikeda, K., Horie-Inoue, K., & Inoue, S. (2020). Roles of Splicing Factors in Hormone-Related Cancer Progression. International Journal of Molecular Sciences, 21(5), 1551. https://doi.org/10.3390/ijms21051551