Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HSP90 and PI3Kα Are Significantly Overexpressed in Melanoma with Positive Correlationship
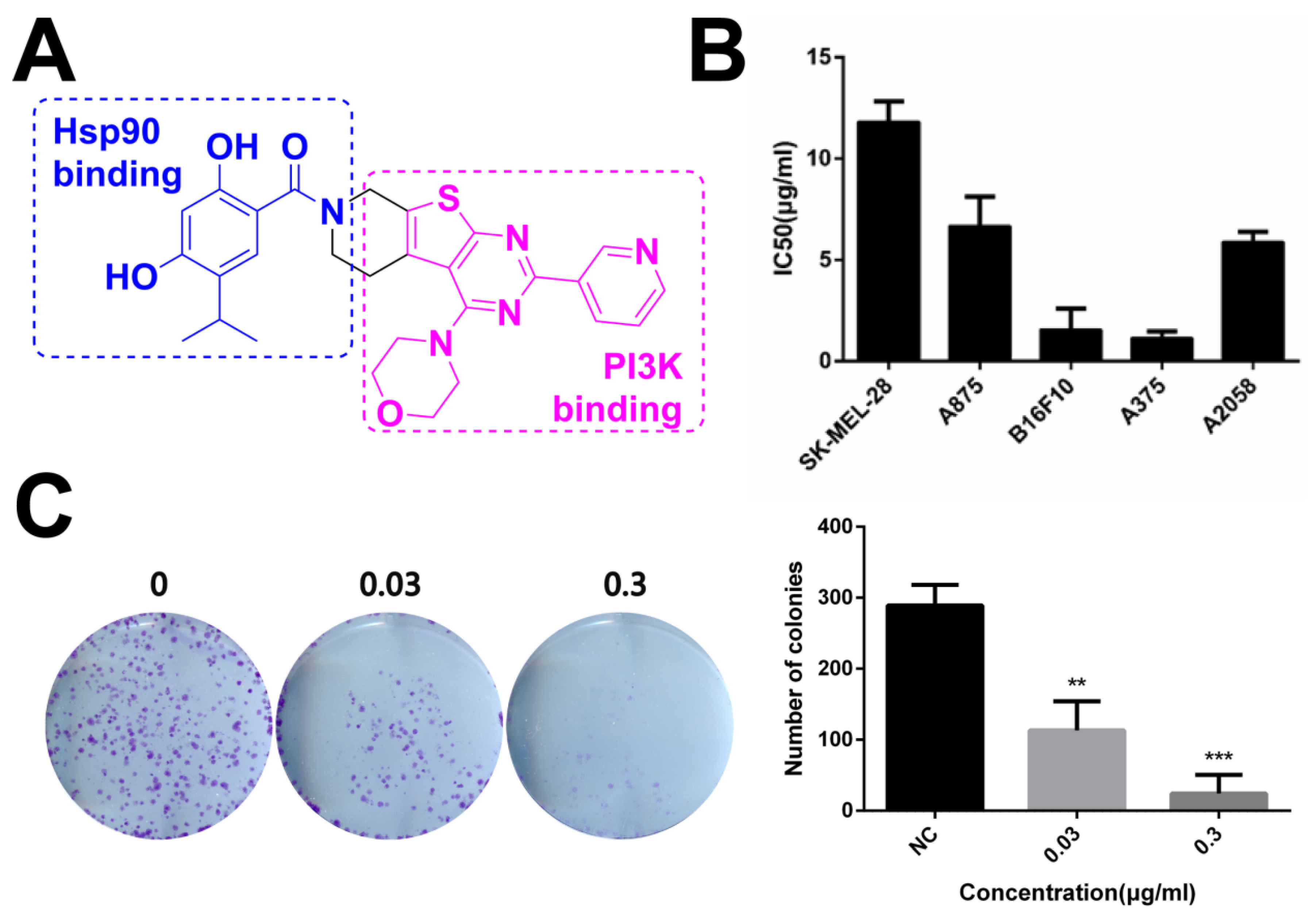
2.2. Novel HSP90/PI3Ka Dual Inhibitor DHP1808 Suppresses A375 Cell Proliferation and Induces Cell Death
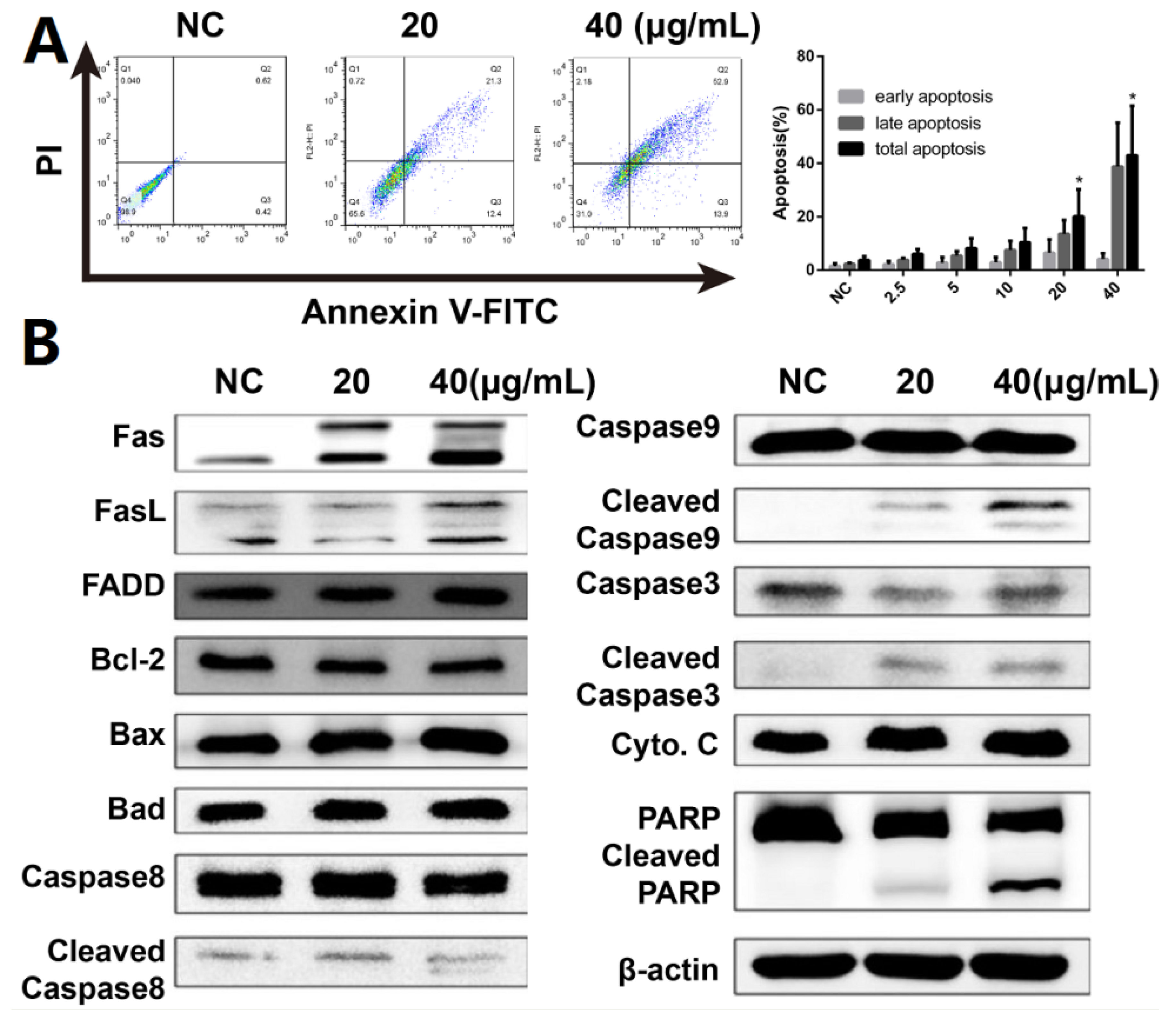
2.3. DHP1808 Induces A375 Cell Apoptosis by Activating the Fas/FasL Signaling Pathway
2.4. DHP1808 Induces Cell Cycle Arrest and Inhibits A375 Cell Migration and Invasion
2.5. DHP1808 Inhibits the Interaction between HSP90 and EGFR
2.6. DHP1808 Induces the PI3K/Akt Signaling Pathway and Regulate the Signaling Pathways Related to HSP90 Client Proteins
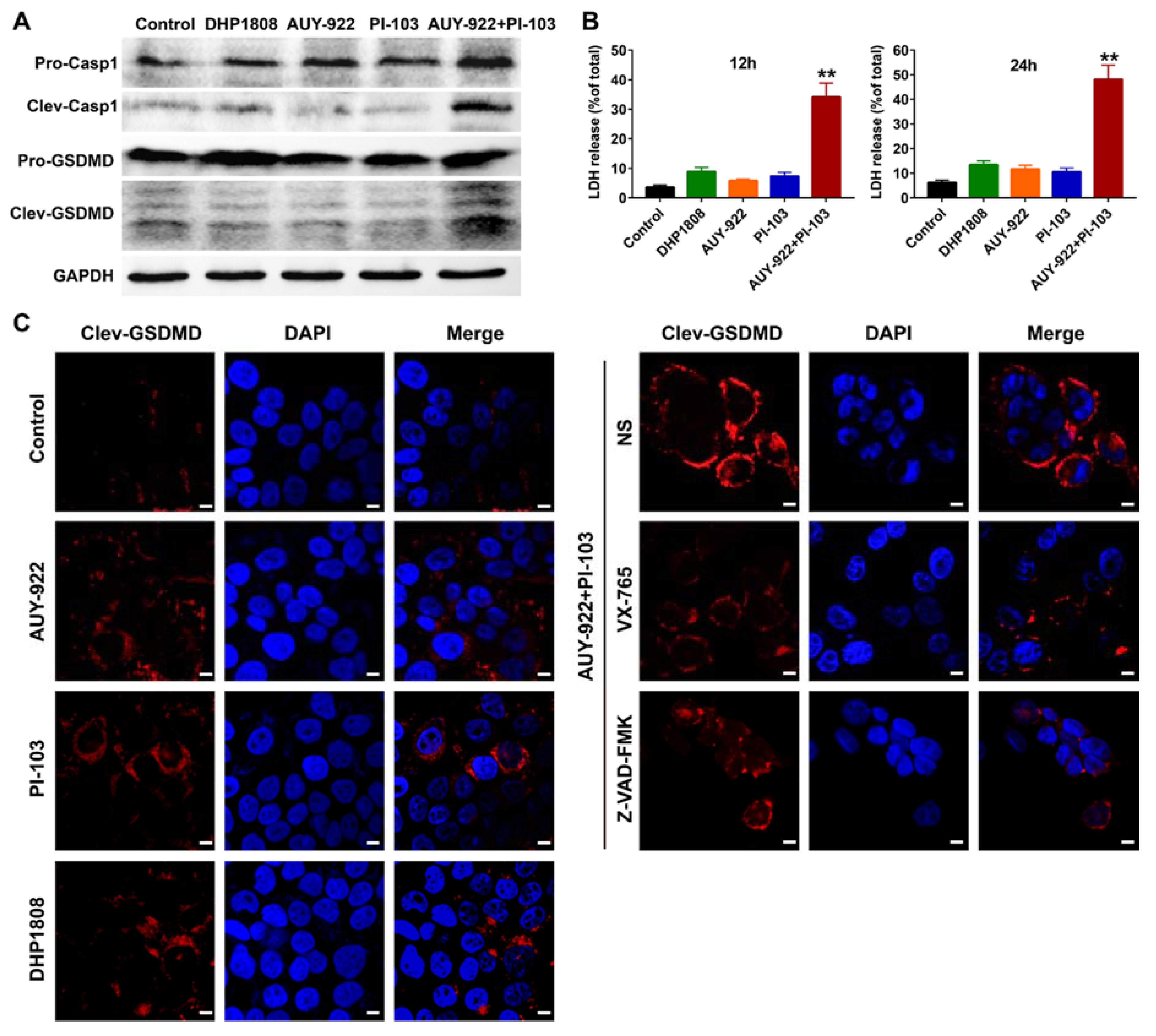
2.7. DHP1808 Resulted in Less Pyroptosis than the Combination of HSP90 and PI3K Inhibitors
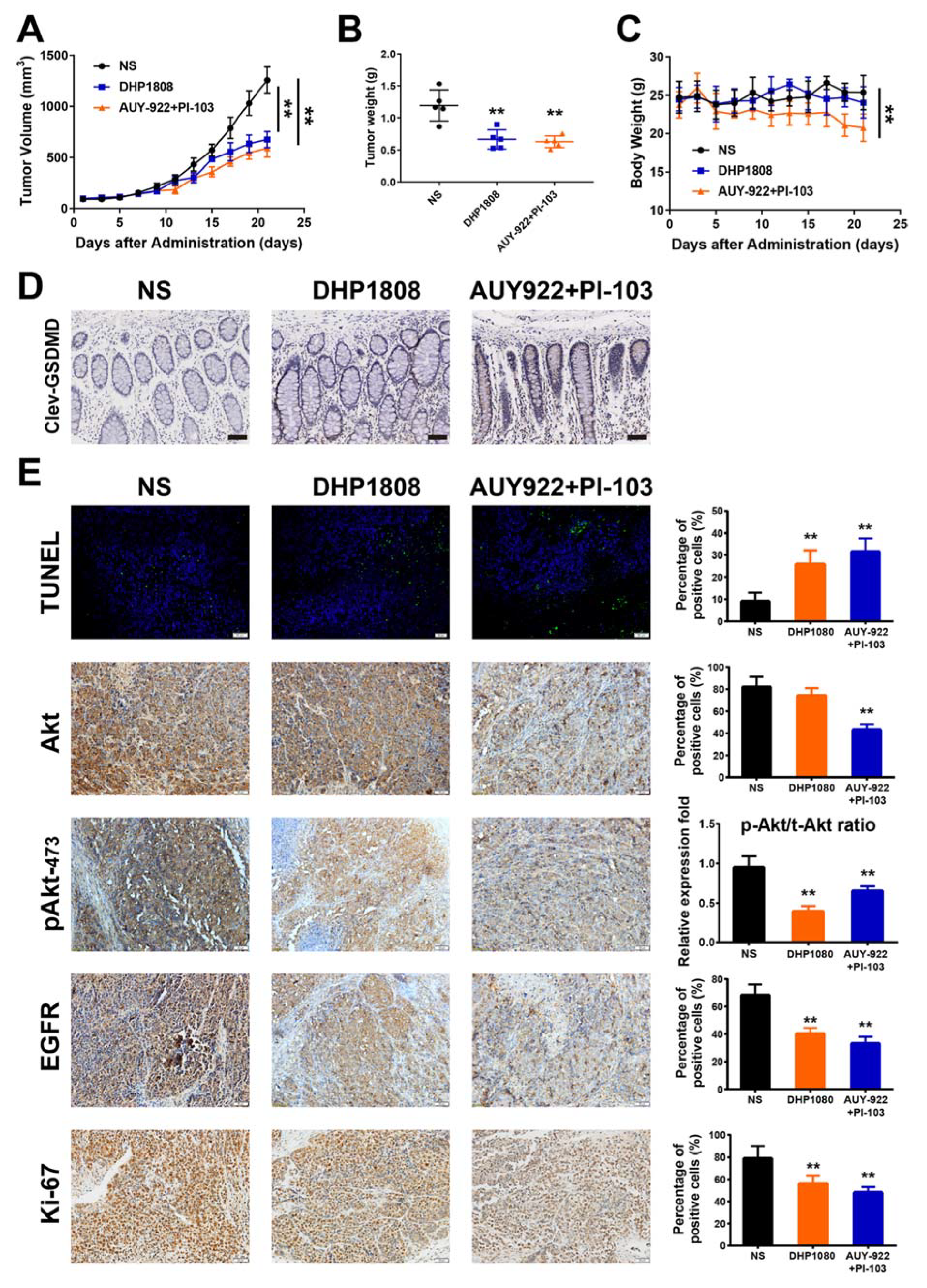
2.8. DHP1808 Inhibited Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Cell Viability Assay
4.3. Western Blotting Analysis
4.4. Immunohistochemistry and Immunofluorescent Assays
4.5. Protein co-IP Assay
4.6. Animal Models
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MAPK | mitogen-activated protein kinasem |
| TOR | the mammalian target of rapamycin |
| Hsp90 | heat shock protein 90 |
| PARP | poly ADP-ribose polymerase |
| FADD | Fas-associating protein with a novel death domain |
| Bcl-2 | B-cell lymphoma-2 |
| EGFR | epidermal growth factor receptor |
| Akt | protein kinase B |
| MDM2 | murine double minute2 |
| ERK | extracellular signal-regulated kinase |
| GSDME | gasdermin E |
| IHC | Immunohistochemistry |
| JNK | c-Jun N-terminal kinase |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide |
| EDTA | ethylenediaminetetraacetic acid |
| IF | immunofluorescent |
| SPF | specific pathogen-free |
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Chen, Y.J.; Yuan, F.J.; Jiang, X.; Lv, Q.; Luo, N.; Gong, C.Y.; Wang, C.T.; Yang, L.; He, G. Discovery of a self-assembling and self-adjuvant lipopeptide as a saccharide-free peptide vaccine targeting EGFRvIII positive cutaneous melanoma. Biomater. Sci. UK 2018, 6, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, S.J.; Tiffen, J.C.; Hersey, P. Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers 2015, 7, 1959–1982. [Google Scholar] [CrossRef] [PubMed]
- Torres-Collado, A.X.; Knott, J.; Jazirehi, A.R. Reversal of Resistance in Targeted Therapy of Metastatic Melanoma: Lessons Learned from Vemurafenib (BRAF(V600E)-Specific Inhibitor). Cancers 2018, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Tripp, M.K.; Watson, M.; Balk, S.J.; Swetter, S.M.; Gershenwald, J.E. State of the Science on Prevention and Screening to Reduce Melanoma Incidence and Mortality: The Time Is Now. CA Cancer J. Clin. 2016, 66, 461–480. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Li, S.J.; Zhang, P.; Wang, Y.J.; Wang, C.T.; Bai, D.; Jiang, X. EMP2 acts as a suppressor of melanoma and is negatively regulated by mTOR-mediated autophagy. J. Cancer 2019, 10, 3582–3592. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.E.; Liu, X.W.; Yang, J.Q.; Zhang, M.; Jin, H.Y.; Ma, X.L.; Shi, H.B. Combination of Immunotherapy With Targeted Therapy: Theory and Practice in Metastatic Melanoma. Front. Immunol. 2019, 10, 990. [Google Scholar] [CrossRef] [Green Version]
- Bollag, G.; Tsai, J.; Zhang, J.Z.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (ST1571, Imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Menzies, A.M.; Long, G.V.; Murali, R. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2012, 6, 391–405. [Google Scholar]
- Sinha, R.; Edmonds, K.; Newton-Bishop, J.A.; Gore, M.E.; Larkin, J.; Fearfield, L. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: Practical advice on diagnosis, prevention and management of the main treatment-related skin toxicities. Br. J. Dermatol. 2012, 167, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Manley, P.; Mestan, J.; Cowan-Jacob, S.; Ray, A.; Griffin, J.D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br. J. Cancer 2006, 94, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.D.; Ozsan, I.; Ospina, S.R.; Gulick, D.; Blair, L.J. Hsp90 Heterocomplexes Regulate Steroid Hormone Receptors: From Stress Response to Psychiatric Disease. Int. J. Mol. Sci. 2019, 20, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.T.; Somogyvari, M.; Soti, C. Hsp90 Stabilizes SIRT1 Orthologs in Mammalian Cells and C-elegans. Int. J. Mol. Sci. 2018, 19, 3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Sidera, K.; Patsavoudi, E. HSP90 Inhibitors: Current Development and Potential in Cancer Therapy. Recent Patents Anti-Cancer 2014, 9, 1–20. [Google Scholar] [CrossRef]
- Liu, K.S.; Liu, H.; Qi, J.H.; Liu, Q.Y.; Liu, Z.; Xia, M.; Xing, G.W.; Wang, S.X.; Wang, Y.F. SNX-2112, an Hsp90 inhibitor, induces apoptosis and autophagy via degradation of Hsp90 client proteins in human melanoma A-375 cells. Cancer Lett. 2012, 318, 180–188. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Rotoloni, C.L.; LaRosa, J.M.; Porter, M.J.; Kelly, L.A.; Nega, K.; Wolfson, E.; Komastu, Y.; Kosovec, J.E.; Kasi, P.M.; Hoppo, T.; et al. Enhanced efficacy of cisplatin and 5-fluorouracil combination with AUY-922 in esophageal adenocarcinoma cells. Cancer Res. 2013, 73, A48. [Google Scholar]
- Amancio, C.; Carmen, B.A.; Oliver, R.; Wolfgang, L.; Juan, F.M.L. The PTEN/PI3K/AKT Signalling Pathway in Cancer, Therapeutic Implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar]
- Georgescu, M.-M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.C.; Sala, V.; Martini, M.; Ferrero, G.B.; Hirsch, E. PI3K Signaling in Tissue Hyper-Proliferation: From Overgrowth Syndromes to Kidney Cysts. Cancers 2017, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.C.; Chen, Y.L.; Lin, P.Y.; Chuang, W.L. Ursolic Acid-Induced Apoptosis via Regulation of the PI3K/Akt and MAPK Signaling Pathways in Huh-7 Cells. Molecules 2018, 23, 2016. [Google Scholar] [CrossRef] [Green Version]
- Neoh, C.A.; Wu, W.T.; Dai, G.F.; Su, J.H.; Liu, C.I.; Su, T.R.; Wu, Y.J. Flaccidoxide-13-Acetate Extracted from the Soft Coral Cladiella kashmani Reduces Human Bladder Cancer Cell Migration and Invasion through Reducing Activation of the FAK/PI3K/AKT/mTOR Signaling Pathway. Molecules 2018, 23, 58. [Google Scholar] [CrossRef] [Green Version]
- Shankar, E.; Weis, M.C.; Avva, J.; Shukla, S.; Shukla, M.; Sreenath, S.N.; Gupta, S. Complex Systems Biology Approach in Connecting PI3K-Akt and NF-kappa B Pathways in Prostate Cancer. Cells 2019, 8, 201. [Google Scholar] [CrossRef] [Green Version]
- Wojtas, B.; Gielniewski, B.; Wojnicki, K.; Maleszewska, M.; Mondal, S.S.; Nauman, P.; Grajkowska, W.; Glass, R.; Schuller, U.; Herold-Mende, C.; et al. Gliosarcoma Is Driven by Alterations in PI3K/Akt, RAS/MAPK Pathways and Characterized by Collagen Gene Expression Signature. Cancers 2019, 11, 284. [Google Scholar] [CrossRef] [Green Version]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Cerami, E.; Gao, J.J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Meyerson, M.; Aldape, K.; Bigner, D.; Mikkelsen, T.; VandenBerg, S.; Kahn, A.; Penny, R.; Ferguson, M.L.; Gerhard, D.S.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Gao, J.J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.C.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.F.; Li, C.W.; Kang, B.X.; Gao, G.; Li, C.; Zhang, Z.M. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunna, N.R.; Bandaru, S.; Akare, U.R.; Rajadhyax, S.; Gutlapalli, V.R.; Yadav, M.; Nayarisseri, A. Multiclass Comparative Virtual Screening to Identify Novel Hsp90 Inhibitors: A Therapeutic Breast Cancer Drug Target. Curr. Top. Med. Chem. 2015, 15, 57–64. [Google Scholar] [CrossRef]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Chapuis, N.; Bardet, V.; Tamburini, J.; Gallay, N.; Willems, L.; Knight, Z.A.; Shokat, K.M.; Azar, N.; Viguie, F.; et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008, 22, 1698–1706. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.F.; Wang, Y.L.; Jiang, X.; Wang, Y.J.; Zhang, N.; Wen, X.; Wang, L.; Jiang, Q.L.; He, G. Design, synthesis and molecular mechanisms of novel dual inhibitors of heat shock protein 90/phosphoinositide 3-kinase alpha (Hsp90/PI3K alpha) against cutaneous melanoma. J. Enzym. Inhib. Med. Chem. 2019, 34, 909–926. [Google Scholar] [CrossRef] [Green Version]
- Raynaud, F.I.; Eccles, S.A.; Patel, S.; Alix, S.; Box, G.; Chuckowree, I.; Folkes, A.; Gowan, S.; Brandon, A.D.; Di Stefano, F.; et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: From PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol. Cancer Ther. 2009, 8, 1725–1738. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.D.; Han, C.Y.; Reid, C.; Kim, S.O. HDAC8-Mediated Epigenetic Reprogramming Plays a Key Role in Resistance to Anthrax Lethal Toxin-Induced Pyroptosis in Macrophages. J. Immunol. 2014, 193, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Ke, B.W.; Tian, M.; Li, J.J.; Liu, B.; He, G. Targeting Programmed Cell Death Using Small-Molecule Compounds to Improve Potential Cancer Therapy. Med. Res. Rev. 2016, 36, 983–1035. [Google Scholar] [CrossRef]
- O’Meara, T.R.; Veri, A.O.; Polvi, E.J.; Li, X.L.; Valaei, S.F.; Diezmann, S.; Cowen, L.E. Mapping the Hsp90 Genetic Network Reveals Ergosterol Biosynthesis and Phosphatidylinositol-4-Kinase Signaling as Core Circuitry Governing Cellular Stress. PLoS Genet. 2016, 12, e1006142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Chen, Y.; Wang, X.Y.; Lu, R.F.; Zhang, S.Y.; Tian, M.; Xie, T.; Liu, B.; He, G. Polygonatum odoratum lectin induces apoptosis and autophagy via targeting EGFR-mediated Ras-Raf-MEK-ERK pathway in human MCF-7 breast cancer cells. Phytomedicine 2014, 21, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Zhou, Z.G.; Rajasingh, S.; Panda, A.; Sampath, V.; Rajasingh, J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int. J. Biochem. Cell B 2018, 102, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Wen, X.; Hao, D.; Zhou, M.K.; Li, X.X.; He, G.; Jiang, X. Insights into autophagy machinery in cells related to skin diseases and strategies for therapeutic modulation. Biomed. Pharmacother. 2019, 113, 108775. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.Q.; Hu, L.C.; Chen, P.D.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J.H. Gasdermin D is an executor of pyroptosis and required for interleukin-1 beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.B.; Ruan, J.B.; Pan, Y.D.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Mai, F.Y.; He, P.; Ye, J.Z.; Xu, L.H.; Ouyang, D.Y.; Li, C.G.; Zeng, Q.Z.; Zeng, C.Y.; Zhang, C.C.; He, X.H.; et al. Caspase-3-mediated GSDME activation contributes to cisplatin- and doxorubicin-induced secondary necrosis in mouse macrophages. Cell Prolif. 2019, 52, e12663. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.J.; Zhao, Y.; Wang, K.; Shi, X.Y.; Wang, Y.; Huang, H.W.; Zhuang, Y.H.; Cai, T.; Wang, F.C.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Zeng, Z.L.; Li, G.H.; Wu, S.Y.; Wang, Z. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2018, 52, e12563. [Google Scholar]
- Chen, N.; Ou, Z.B.; Zhang, W.F.; Zhu, X.W.; Li, P.Z.; Gong, J.P. Cathepsin B regulates non-canonical NLRP3 inflammasome pathway by modulating activation of caspase-11 in Kupffer cells. Cell Prolif. 2018, 51, e12487. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; He, W.T.; Hu, L.C.; Li, J.X.; Fang, Y.; Wang, X.; Xu, X.Z.; Wang, Z.; Huang, K.; Han, J.H. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Gao, W.Q.; Shi, X.Y.; Ding, J.J.; Liu, W.; He, H.B.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.A.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-Kinase Hyperactivation Results in Lapatinib Resistance that Is Reversed by the mTOR/Phosphatidylinositol 3-Kinase Inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.H.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.C.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.A.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Busconi, L.; Guan, J.Z.; Denker, B.M. Degradation of heterotrimeric G alpha(o) subunits via the proteosome pathway is induced by the hsp90-specific compound geldanamycin. J. Biol. Chem. 2000, 275, 1565–1569. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.G.; Moon, J.S.; Kim, E.J.; Lee, S.H.; Oh, J.W. Destabilization of PDK1 by Hsp90 inactivation suppresses hepatitis C virus replication through inhibition of PRK2-mediated viral RNA polymerase phosphorylation. Biochem. Biophys. Res. Commun. 2012, 421, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Mabjeesh, N.J.; Post, D.E.; Willard, M.T.; Kaur, B.; Van Meir, E.G.; Simons, J.W.; Zhong, H. Geldanamycin induces degradation of hypoxia-inducible factor 1 alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002, 62, 2478–2482. [Google Scholar] [PubMed]
- Lage, H.; Helmbach, H.; Grottke, C.; Dietel, M.; Schadendorf, D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001, 494, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.Y.; Zhang, W.D.; Cheng, C.; Mo, F.B.; Chen, L.; Peng, G.F.; Cai, X.Y.; Wang, J.L.; Yang, S.H.; Liu, X.Z. Dioscin inhibits the growth of human osteosarcoma by inducing G2/M-phase arrest, apoptosis, and GSDME-dependent cell death in vitro and in vivo. J. Cell. Physiol. 2020, 235, 2911–2924. [Google Scholar] [CrossRef]
- Hu, J.; Dong, Y.; Ding, L.; Dong, Y.; Wu, Z.H.; Wang, W.P.; Shen, M.; Duan, Y.R. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct. Target. Ther. 2019, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.L.; He, S.D. GSDME as an executioner of chemotherapy-induced cell death. Sci. China Life Sci. 2017, 60, 1291–1294. [Google Scholar] [CrossRef]
- Zhu, H.P.; Xie, K.; He, X.-H.; Huang, W.; Zeng, R.; Fan, Y.; Peng, C.; He, G.; Han, B. Organocatalytic diastereoselective [3 + 2] cyclization of MBH carbonates with dinucleophiles: Synthesis of bicyclic imidazoline derivatives that inhibit MDM2–p53 interaction. Chem. Commun. 2019, 55, 11374–11377. [Google Scholar] [CrossRef]
- Wang, T.; Su, X.; Fang, J.; Xin, X.; Zhao, X.; Gaur, U.M.A.; Wen, Q.; Xu, J.; Littlt, P.J.; Zheng, W. Tanshinone IIA Attenuates Insulin Like Growth Factor 1 -Induced Cell Proliferation in PC12 Cells through the PI3K/Akt and MEK/ERK Pathways. Int. J. Mol. Sci. 2018, 19, 2719. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo Genome Editing as a Therapeutic Approach. Int. J. Mol. Sci. 2018, 19, 2739. [Google Scholar] [CrossRef] [Green Version]
- Hao, D.; Wen, X.; Liu, L.; Wang, L.; Zhou, X.; Li, Y.; Zeng, X.; He, G.; Jiang, X. Sanshool improves UVB-induced skin photodamage by targeting JAK2/STAT3-dependent autophagy. Cell Death Dis. 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, C.; Huang, W.; Haruehanroengra, P.; Peng, C.; Sheng, J.; Han, B.; He, G. Application of organocatalysis in bioorganometallic chemistry: Asymmetric synthesis of multifunctionalized spirocyclic pyrazolone–ferrocene hybrids as novel RalA inhibitors. Org. Chem. Front. 2018, 5, 2229–2233. [Google Scholar] [CrossRef]
- Zhao, Q.; Peng, C.; Huang, H.; Liu, S.J.; Zhong, Y.J.; Huang, W.; He, G.; Han, B. Asymmetric synthesis of tetrahydroisoquinoline-fused spirooxindoles as Ras-GTP inhibitors that inhibit colon adenocarcinoma cell proliferation and invasion. Chem. Commun. 2018, 54, 8359–8362. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Chen, Y.; Liu, J.; Jiang, Q.; Yang, S.; Guo, L.; He, G. Design, synthesis, and biological evaluation of polo-like kinase 1/eukaryotic elongation factor 2 kinase (PLK1/EEF2K) dual inhibitors for regulating breast cancer cells apoptosis and autophagy. Eur. J. Med. Chem. 2018, 144, 517–528. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Zhu, H.-P.; Xie, X.; Mao, Q.; Liu, Y.-Q.; He, X.-H.; Peng, C.; Jiang, Q.-L.; Huang, W. Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 1845. https://doi.org/10.3390/ijms21051845
Zhao Q, Zhu H-P, Xie X, Mao Q, Liu Y-Q, He X-H, Peng C, Jiang Q-L, Huang W. Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways. International Journal of Molecular Sciences. 2020; 21(5):1845. https://doi.org/10.3390/ijms21051845
Chicago/Turabian StyleZhao, Qian, Hong-Ping Zhu, Xin Xie, Qing Mao, Yan-Qing Liu, Xiang-Hong He, Cheng Peng, Qing-Lin Jiang, and Wei Huang. 2020. "Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways" International Journal of Molecular Sciences 21, no. 5: 1845. https://doi.org/10.3390/ijms21051845
APA StyleZhao, Q., Zhu, H. -P., Xie, X., Mao, Q., Liu, Y. -Q., He, X. -H., Peng, C., Jiang, Q. -L., & Huang, W. (2020). Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways. International Journal of Molecular Sciences, 21(5), 1845. https://doi.org/10.3390/ijms21051845