Application of an Inter-Species Extrapolation Method for the Prediction of Drug Interactions between Propolis and Duloxetine in Humans

 , ,
, ,  ,
, 
Abstract
:
1. Introduction
2. Results
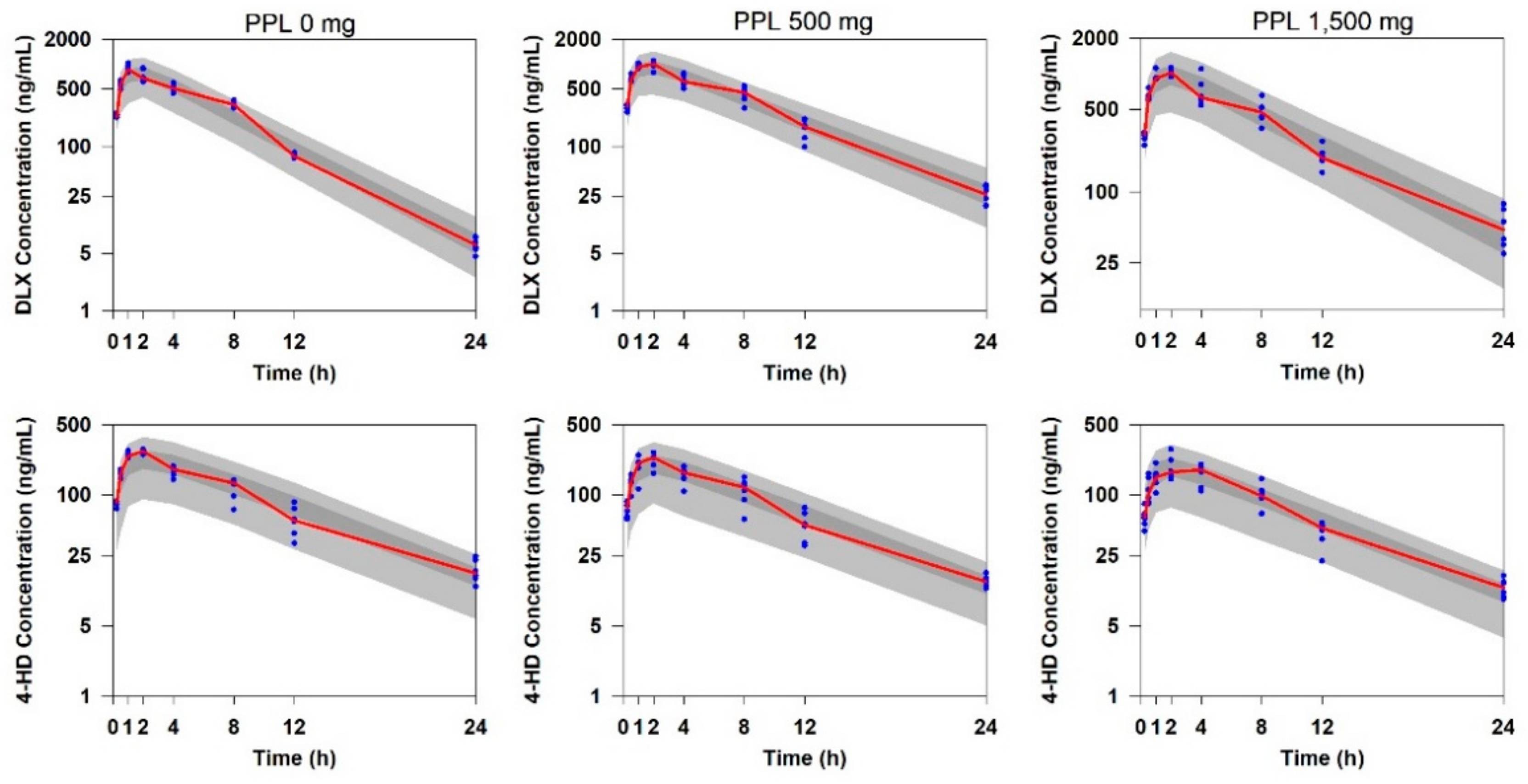
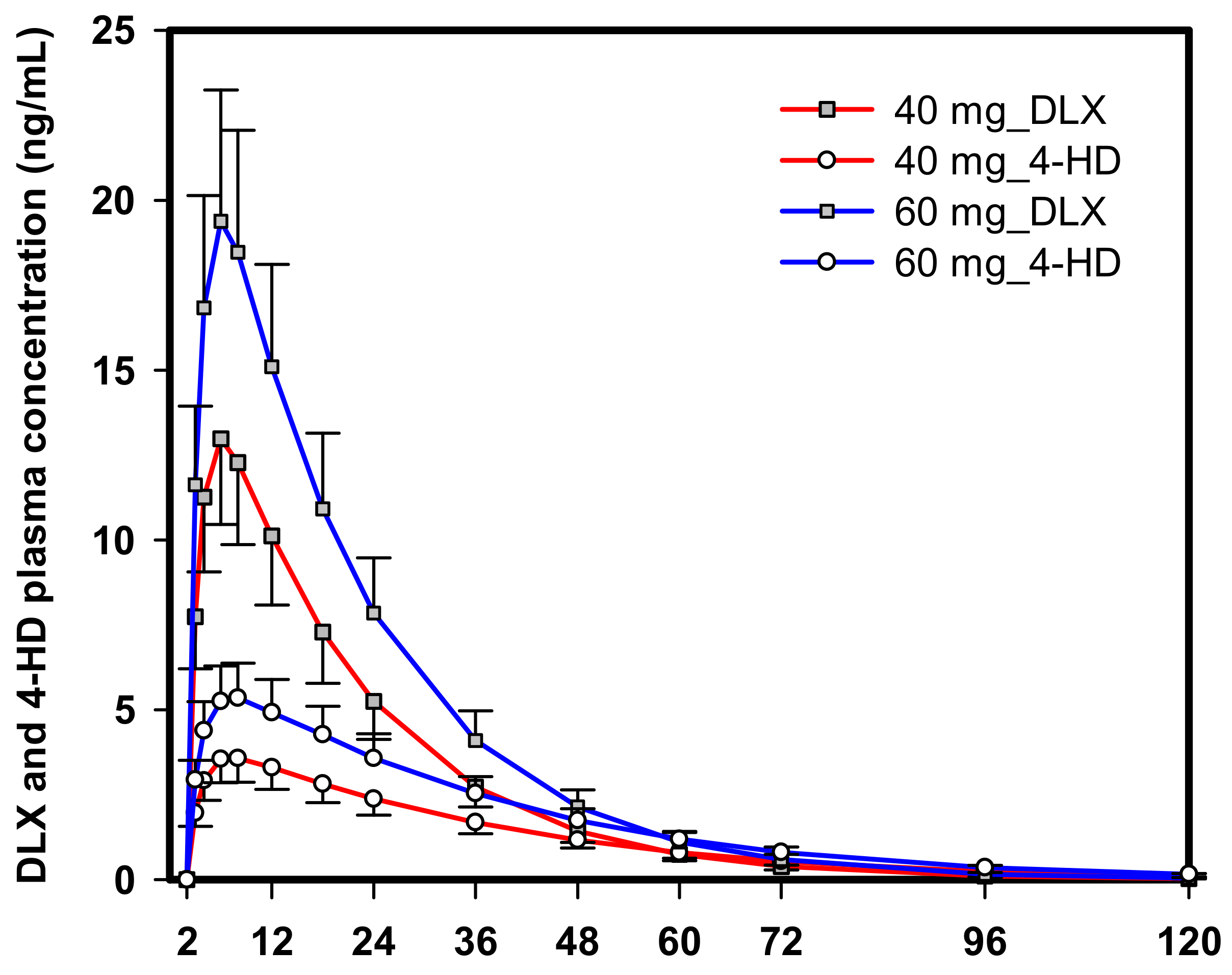
2.1. PK Data of DLX and 4-HD in Rats
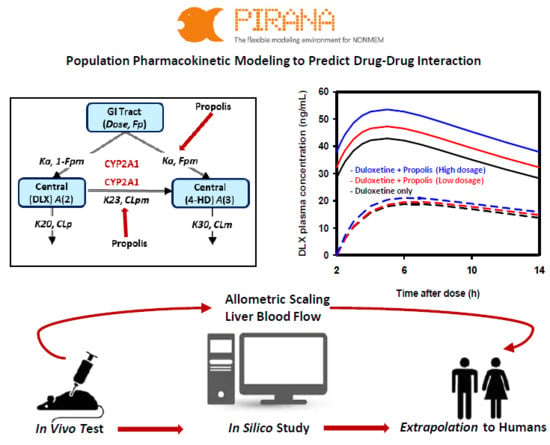
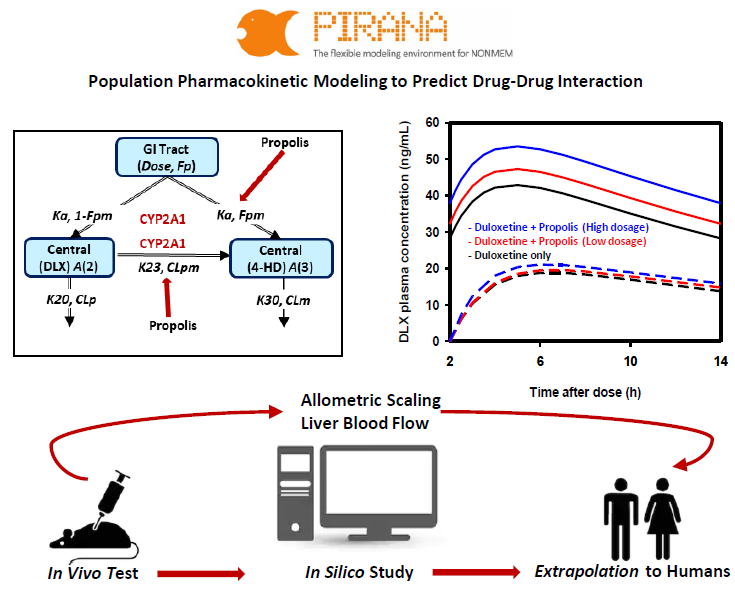
2.2. Population PK Model for DLX and 4-HD in Rats
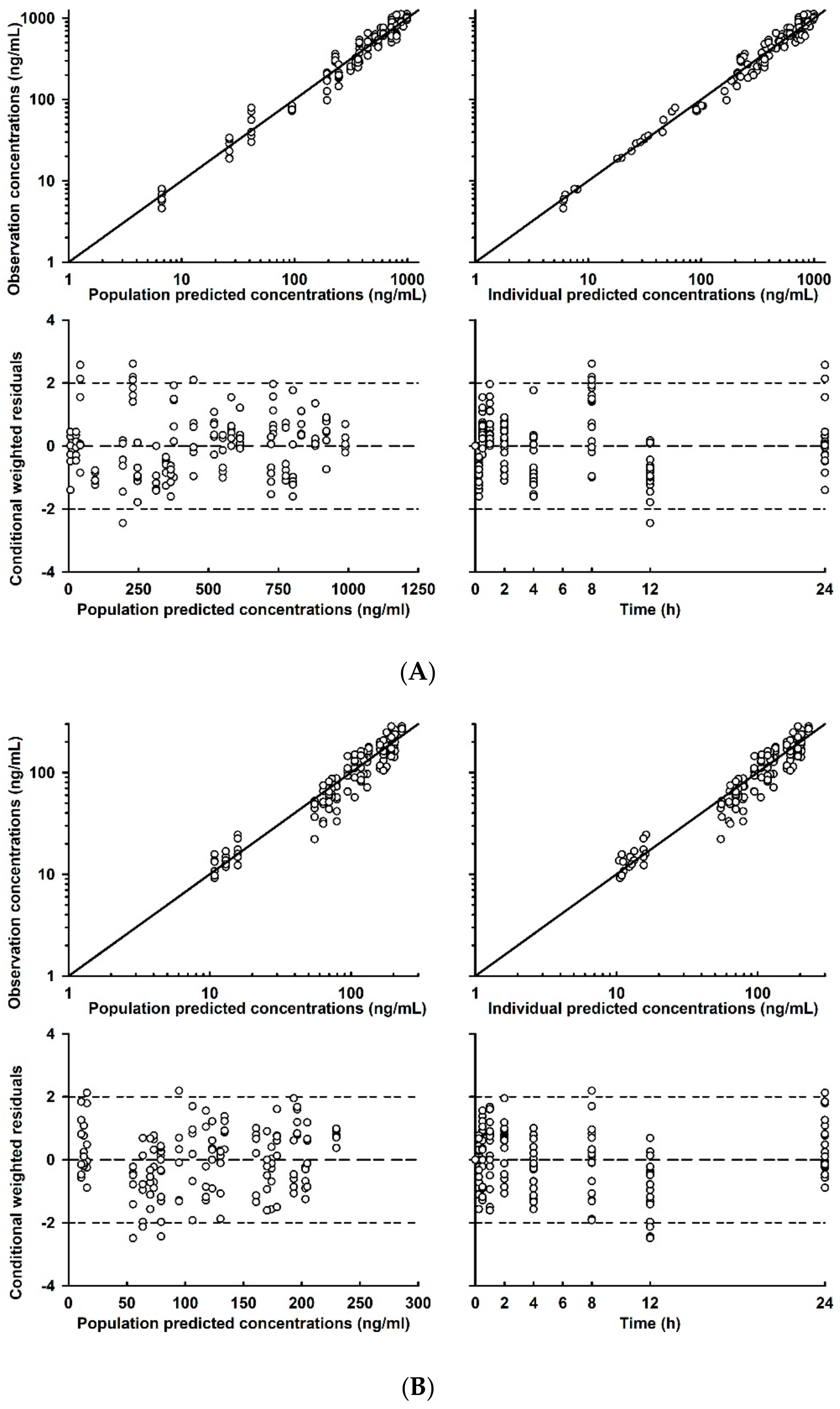
2.3. Population PK Model Evaluation
2.4. Extrapolation of PKs of DLX and 4-HD in Humans
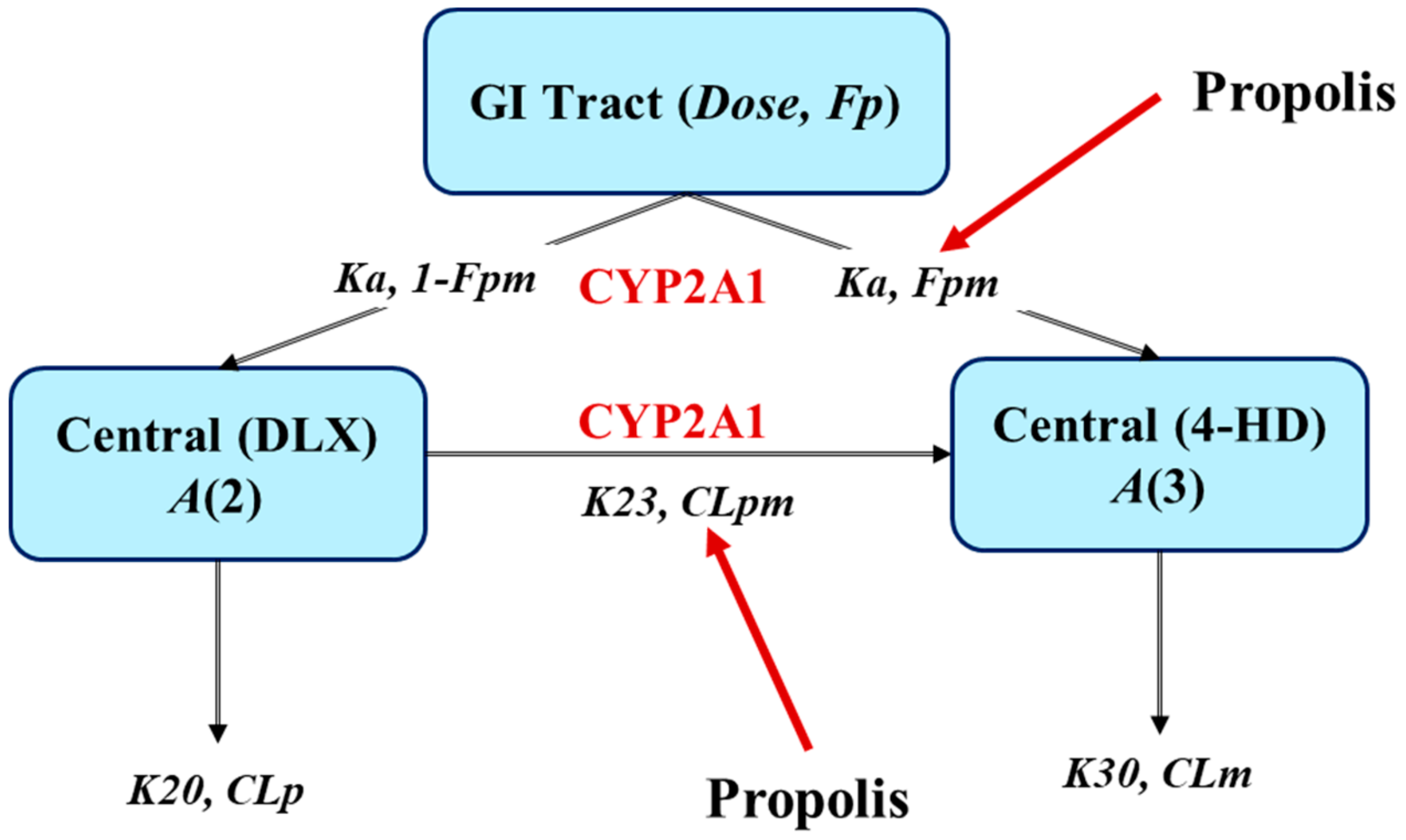
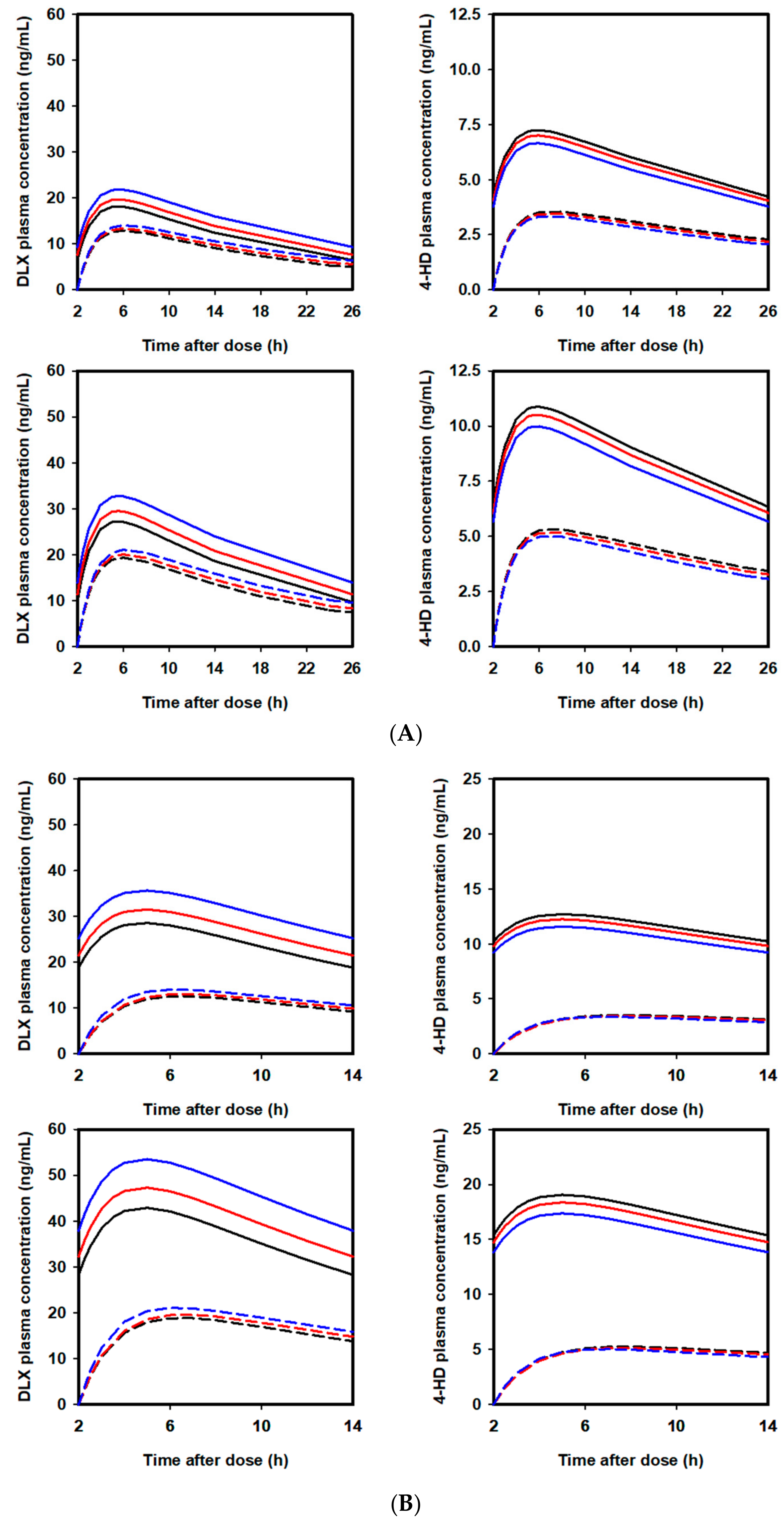
2.5. Extrapolation of Effect of PPL on PKs of DLX in Humans
3. Discussions
4. Material and Methods
4.1. Quantitative Determination of Major Ingredients in Propolis Extract
4.2. PK Study Design in Rats and Data Analysis
4.3. Rat Population PK Model Development
4.4. Rat Population PK Model Evaluation
4.5. Extrapolating Population PK of DLX in Humans
4.6. Simulation of PPL Effects on PKs of DLX in Humans
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ghisalberti, E.L. Propolis: A Review. Bee World 1979, 60, 59–84. [Google Scholar] [CrossRef]
- Castaldo, S.; Capasso, F. Propolis, an old remedy used in modern medicine. Fitoterapia 2002, 73, S1–S6. [Google Scholar] [CrossRef]
- Paulino, N.; Teixeira, C.; Martins, R.; Scremin, A.; Dirsch, V.; Vollmar, A.; Abreu, S.; de Castro, S.; Marcucci, M. Evaluation of the Analgesic and Anti-Inflammatory Effects of a Brazilian Green Propolis. Planta Med. 2006, 72, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-khawaga, O.-A.Y.; Salem, T.A.; Elshal, M.F. Protective role of Egyptian propolis against tumor in mice. Clin. Chim. Acta 2003, 338, 11–16. [Google Scholar] [CrossRef]
- Ozkul, Y.; Silici, S.; Eroğlu, E. The anticarcinogenic effect of propolis in human lymphocytes culture. Phytomedicine 2005, 12, 742–747. [Google Scholar] [CrossRef]
- Ahn, M.-R.; Kunimasa, K.; Kumazawa, S.; Nakayama, T.; Kaji, K.; Uto, Y.; Hori, H.; Nagasawa, H.; Ohta, T. Correlation between antiangiogenic activity and antioxidant activity of various components from propolis. Mol. Nutr. Food Res. 2009, 53, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.; Ismail, S.; Rohin, M.; Harun, A.; Aziz, A.; Haque, M. Antifungal Properties of Malaysian Tualang Honey and Stingless Bee Propolis against Candida Albicans and Cryptococcus Neoformans. J. Appl. Pharm. Sci. 2016, 044–050. [Google Scholar] [CrossRef] [Green Version]
- Kartal, M.; Yıldız, S.; Kaya, S.; Kurucu, S.; Topçu, G. Antimicrobial activity of propolis samples from two different regions of Anatolia. J. Ethnopharmacol. 2003, 86, 69–73. [Google Scholar] [CrossRef]
- Amoros, M.; Lurton, E.; Boustie, J.; Girre, L.; Sauvager, F.; Cormier, M. Comparison of the Anti-Herpes Simplex Virus Activities of Propolis and 3-Methyl-but-2-enyl Caffeate. J. Nat. Prod. 1994, 57, 644–647. [Google Scholar] [CrossRef]
- Seo, K.W.; Park, M.; Song, Y.J.; Kim, S.-J.; Yoon, K.R. The protective effects of Propolis on hepatic injury and its mechanism. Phytother. Res. 2003, 17, 250–253. [Google Scholar]
- González, R.; Corcho, I.; Remirez, D.; Rodriguez, S.; Ancheta, O.; Merino, N.; González, A.; Pascual, C. Hepatoprotective effects of propolis extract on carbon tetrachloride-induced liver injury in rats. Phyther. Res. 1995, 9, 114–117. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, Y.H.; Chen, C.F.; Chung, C.Y.; Hsu, S.H. The hepatoprotective and therapeutic effects of propolis ethanol extract on chronic alcohol-induced liver injuries. Am. J. Chin. Med. 1997, 25, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Naramoto, K.; Kato, M.; Ichihara, K. Effects of an Ethanol Extract of Brazilian Green Propolis on Human Cytochrome P450 Enzyme Activities in Vitro. J. Agric. Food Chem. 2014, 62, 11296–11302. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.S.; Oh, S.J.; Oh, J.M.; Lee, J.-Y.; Lee, S.Y.; Chae, J.; Kwon, K.; Kim, S.K. Inhibition of Cytochrome P450 by Propolis in Human Liver Microsomes. Toxicol. Res. 2016, 32, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Sato, Y.; Kumagai, T.; Yoshinari, K.; Nagata, K. Effect of health foods on cytochrome P450-mediated drug metabolism. J. Pharm. Health Care Sci. 2017, 3, 14. [Google Scholar] [CrossRef]
- Šarić Mustapić, D.; Debeljak, Ž.; Maleš, Ž.; Bojić, M. The Inhibitory Effect of Flavonoid Aglycones on the Metabolic Activity of CYP3A4 Enzyme. Molecules 2018, 23, 2553. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Cymbalta-Product Information; European Medicines Agency: Amsterdam, The Netherlands, 2004. [Google Scholar]
- U.S. Food and Drug Administration. Cymbalta-Product Information; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2004. [Google Scholar]
- Lantz, R.J.; Gillespie, T.A.; Rash, T.J.; Kuo, F.; Skinner, M.; Kuan, H.-Y.; Knadler, M.P. Metabolism, excretion, and pharmacokinetics of duloxetine in healthy human subjects. Drug Metab. Dispos. 2003, 31, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Lobo, E.D.; Bergstrom, R.F.; Reddy, S.; Quinlan, T.; Chappell, J.; Hong, Q.; Ring, B.; Knadler, M.P. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharmacokinet. 2008, 47, 191–202. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications Guidance for Industry; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- European Medicines Agency Guideline on the Investigation of Drug Interactions; European Medicines Agency: Amsterdam, The Netherlands, 2012.
- Boxenbaum, H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef]
- Tang, H.; Hussain, A.; Leal, M.; Mayersohn, M.; Fluhler, E. Interspecies Prediction of Human Drug Clearance Based on Scaling Data from One or Two Animal Species. Drug Metab. Dispos. 2007, 35, 1886–1893. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, G.W.; Masucci, J.A.; Yan, Z.; Hageman, W. Allometric scaling of pharmacokinetic parameters in drug discovery: Can human CL, Vss and t1/2 be predicted fromin-vivo rat data? Eur. J. Drug Metab. Pharmacokinet. 2004, 29, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.L. Animal scale-up. J. Pharmacokinet. Biopharm. 1973, 1, 435–461. [Google Scholar] [CrossRef]
- Ward, K.W.; Smith, B.R. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. I. Clearance. Drug Metab. Dispos. 2004, 32, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagilla, R.; Ward, K.W. A comprehensive analysis of the role of correction factors in the allometric predictivity of clearance from rat, dog, and monkey to humans. J. Pharm. Sci. 2004, 93, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration Bioanalytical Method Validation: Guidance for Industry. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 15 June 2018).
- Huang, S.-M.; Temple, R.; Throckmorton, D.C.; Lesko, L.J. Drug Interaction Studies: Study Design, Data Analysis, and Implications for Dosing and Labeling. Clin. Pharmacol. Ther. 2007, 81, 298–304. [Google Scholar] [CrossRef]
- Palleria, C.; Di Paolo, A.; Giofrè, C.; Caglioti, C.; Leuzzi, G.; Siniscalchi, A.; De Sarro, G.; Gallelli, L. Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 2013, 18, 601–610. [Google Scholar]
- Kim, S.H.; Kim, W.B.; Lee, M.G. Interspecies pharmacokinetic scaling of a new carbapenem, DA-1131, in mice, rats, rabbits and dogs, and prediction of human pharmacokinetics. Biopharm. Drug Dispos. 1998, 19, 231–235. [Google Scholar] [CrossRef]
- Shim, H.J.; Kim, Y.C.; Lee, J.H.; Kwon, J.W.; Kim, W.B.; Kim, Y.G.; Kim, S.H.; Lee, M.G. Interspecies pharmacokinetic scaling of DA-8159, a new erectogenic, in mice, rats, rabbits and dogs, and prediction of human pharmacokinetics. Biopharm. Drug Dispos. 2005, 26, 269–277. [Google Scholar] [CrossRef]
- Lave, T.; Dupin, S.; Schmitt, M.; Kapps, M.; Meyer, J.; Morgenroth, B.; Chou, R.C.; Jaeck, D.; Coassolo, P. Interspecies scaling of tolcapone, a new inhibitor of catechol- O -methyltransferase (COMT). Use of in vitro data from hepatocytes to predict metabolic clearance in animals and humans. Xenobiotica 1996, 26, 839–851. [Google Scholar] [CrossRef]
- Mahmood, I.; Balian, J.D. Interspecies Scaling: Predicting Pharmacokinetic Parameters of Antiepileptic Drugs in Humans from Animals with Special Emphasis on Clearance. J. Pharm. Sci. 1996, 85, 411–414. [Google Scholar] [CrossRef]
- Mahmood, I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv. Drug Deliv. Rev. 2007, 59, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Yassen, A.; Olofsen, E.; Kan, J.; Dahan, A.; Danhof, M. Animal-to-Human Extrapolation of the Pharmacokinetic and Pharmacodynamic Properties of Buprenorphine. Clin. Pharmacokinet. 2007, 46, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Gallati, H.; Schiller, C.D. Animal pharmacokinetics of the tumor necrosis factor receptor-immunoglobulin fusion protein lenercept and their extrapolation to humans. Drug Metab. Dispos. 1999, 27, 21–25. [Google Scholar] [PubMed]
- Ward, K.W.; Coon, D.J.; Magiera, D.; Bhadresa, S.; Struharik, M.; Lawrence, M.S. Exploration of the African green monkey as a preclinical pharmacokinetic model: Oral pharmacokinetic parameters and drug-drug interactions. Xenobiotica 2009, 39, 266–272. [Google Scholar] [CrossRef]
- Mahmood, I. Integration of in Vitro Data and Brain Weight in Allometric Scaling to Predict Clearance in Humans: Some Suggestions. J. Pharm. Sci. 1998, 87, 527–529. [Google Scholar] [CrossRef]
- Lavé, T.; Coassolo, P.; Reigner, B. Prediction of hepatic metabolic clearance based on interspecies allometric scaling techniques and in vitro-in vivo correlations. Clin. Pharmacokinet. 1999, 36, 211–231. [Google Scholar]
- Huang, Q.; Riviere, J.E. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Tianmei, S.; Knadler, M.P.; Lim, M.T.; Yeo, K.P.; Teng, L.; Liang, S.; Pan, A.X.; Lobo, E.D. Pharmacokinetics and tolerability of duloxetine following oral administration to healthy Chinese subjects. Clin. Pharmacokinet. 2007, 46, 767–775. [Google Scholar] [CrossRef]
- Lobo, E.D.; Loghin, C.; Knadler, M.P.; Quinlan, T.; Zhang, L.; Chappell, J.; Lucas, R.; Bergstrom, R.F. Pharmacokinetics of duloxetine in breast milk and plasma of healthy postpartum women. Clin. Pharmacokinet. 2008, 47, 103–109. [Google Scholar] [CrossRef]
- Skinner, M. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin. Pharmacol. Ther. 2003, 73, 170–177. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Li, Y.; Shen, Y. Pharmacokinetics and Safety of Duloxetine Enteric-coated Tablets in Chinese Healthy Volunteers: A Randomized, Open-label, Single- and Multiple-dose Study. Clin. Psychopharmacol. Neurosci. 2013, 11, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Yeo, K.P.; Pan, A.X.; Lim, M.; Knadler, M.P.; Small, D.S. Duloxetine pharmacokinetics are similar in Japanese and Caucasian subjects. Br. J. Clin. Pharmacol. 2007, 63, 310–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolivette, L.J.; Ward, K.W. Extrapolation of human pharmacokinetic parameters from rat, dog, and monkey data: Molecular properties associated with extrapolative success or failure. J. Pharm. Sci. 2005, 94, 1467–1483. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.A.; Jolivette, L.J.; Nagilla, R.; Ward, K.W. Extrapolation of preclinical pharmacokinetics and molecular feature analysis of “discovery-like” molecules to predict human pharmacokinetics. Drug Metab. Dispos. 2006, 34, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, M.R.; Lou, X.; Brown, R.R.; Hutchaleelaha, A. Allometric pharmacokinetic scaling: Towards the prediction of human oral pharmacokinetics. Pharm. Res. 2000, 17, 410–418. [Google Scholar] [CrossRef]
- Sharma, A.; Goldberg, M.J.; Cerimele, B.J. Pharmacokinetics and safety of duloxetine, a dual-serotonin and norepinephrine reuptake inhibitor. J. Clin. Pharmacol. 2000, 40, 161–167. [Google Scholar] [CrossRef]
- Ferngrove Pharmaceuticals Australia Pty Ltd. Black Propolis 5000 mg. Available online: https://global.rakuten.com/en/store/the-natural/item/plo5000-100c/ (accessed on 14 April 2019).
- Ever Health EverHealth Propolis 5000, 120 Capsules. Available online: http://www.everhealth.co.nz/portfolio-item/propolis5000/ (accessed on 14 April 2019).
- My Natural Health Limited Health Vibrant Propolis 5000. Available online: https://www.mynaturalhealth.co.nz/products/view/health-vibrant-propolis-5000 (accessed on 25 February 2019).
- True Blue Premium Dark Bee Propolis 5000mg 250 Capsules Australian Made. Available online: https://www.amazon.com/True-Blue-Propolis-Capsules-Australian/dp/B0768SLYK8 (accessed on 15 February 2019).
- Chae, J.; Baek, H.; Kim, S.K.; Kang, H.; Kwon, K. Quantitative determination of duloxetine and its metabolite in rat plasma by HPLC-MS/MS. Biomed. Chromatogr. 2013, 27, 953–955. [Google Scholar] [CrossRef]
- Boeckmann, A.J.; Beal, S.L.; Sheiner, L.B. NONMEM User’s Guide, Part I; NONMEM Project Group, University of California at San Francisco: San Francisco, CA, USA, 1998. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model No. | Model Description | df. | Change in OFV | Compared with | Significance (p-Value) |
|---|---|---|---|---|---|
| 1 | Base model | − | − | − | − |
| 2 | Model 1 with PPL as covariate for Fpm | 1 | −38.4 | Model 1 | <0.000 |
| 3 | Model 1 with PPL as covariate for CLpm | 2 | −28.7 | Model 1 | <0.000 |
| 4 a | Model 1 with PPL as covariate for Fpm and CLp | 3 | −47.8 | Model 1 | <0.000 |
| 4 a | Model 1 with PPL as covariate for Fpm and CLp | 2 | −10.6 | Model 2 | <0.005 |
| Parameters | Unit | Estimates | RSE (%) | Shrinkage (%) | Bootstrap Replicates (n = 1000) | |||
|---|---|---|---|---|---|---|---|---|
| Median | 95% CI | |||||||
| 1/h | 1.35 | 8.10 | 1.35 | 1.24 | − | 1.45 | ||
| Emax_ | 0.147 | 85.7 | 0.198 | 0.0435 | − | 0.844 | ||
| IC50_ | mg/kg | 538 | 358 | 806 | 45.7 | − | 5478 | |
| 0.589 | 15.9 | 0.574 | 0.451 | − | 0.746 | |||
| L/h/kg | 1.97 | 22.6 | 2.10 | 1.08 | − | 2.80 | ||
| L/kg | 14.6 | 26.4 | 15.1 | 9.01 | − | 19.6 | ||
| Emax_ | 1.00 | 1.00 | 1.00 | − | 1.00 | |||
| IC50_ | mg/kg | 276 | 77.2 | 270 | 60.1 | − | 596 | |
| L/h/kg | 1.26 | 30.7 | 1.24 | 0.770 | − | 1.77 | ||
| L/h/kg | 12.3 | 13.6 | 11.9 | 9.45 | − | 16.0 | ||
| L/kg | 84.2 | 21.6 | 81.3 | 64.3 | − | 110 | ||
| Inter-individual variability (IIV, %) | ||||||||
| IIV | 7.90 | 70.0 | 14.0 | 7.10 | 3.21 | − | 10.2 | |
| Residual variability (%) | ||||||||
| Prop_p | 19.9 | 13.4 | 19.8 | 18.0 | − | 21.9 | ||
| Prop_m | 24.0 | 10.3 | 23.5 | 20.8 | − | 26.2 | ||
| Parameters | Unit | Predicted Value |
|---|---|---|
| 1/h | 0.687 | |
| L/h | 34.1 | |
| L/h | 21.8 | |
| L | 1022 | |
| 1/h | 0.0547 | |
| L/h | 212 | |
| L/h | 5894 |
| Scenario | Parameter | PPL 0 mg | PPL 5000 mg | Difference a (%) | PPL 15,000 mg | Difference b (%) | |
|---|---|---|---|---|---|---|---|
| 12.7 ± 0.602 | 13.8 ± 0.699 | 15.3 ± 0.867 | |||||
| Single dose | 40 mg | 12.9 ± 0.0928 | |||||
| 294 ± 13.8 | |||||||
| 60 mg | 19.4 ± 0.139 | ||||||
| 441 ± 20.7 | |||||||
| Multiple dose, once daily | 40 mg | 18.2 ± 0.557 | 19.7 ± 0.671 | 8.35 | 21.9 ± 0.868 | 20.2 | |
| 294 ± 14.0 | 328 ± 16.8 | 11.6 | 376 ± 21.5 | 28.0 | |||
| 60 mg | 27.3 ± 0.836 | 29.6 ± 1.01 | 8.35 | 32.8 ± 1.30 | 20.2 | ||
| 441 ± 21.0 | 492 ± 25.2 | 11.6 | 561 ± 32.3 | 28.0 | |||
| Multiple dose, twice daily | 40 mg | 28.7 ± 1.16 | 31.6 ± 1.39 | 10.2 | 35.7 ± 1.78 | 24.6 | |
| 294 ± 14.0 | 328 ± 16.8 | 11.6 | 376 ± 21.5 | 28.0 | |||
| 60 mg | 43.0 ± 1.74 | 47.4 ± 2.08 | 10.2 | 53.6 ± 2.67 | 24.6 | ||
| 441 ± 21.0 | 492 ± 25.2 | 11.6 | 564 ± 32.3 | 28.0 | |||
| Ingredient | Content (µg/mg) * | ||
|---|---|---|---|
| Chrysin | 23.57 | ± | 3.02 |
| Galangin | 7.45 | ± | 0.51 |
| Kaempferide | 4.30 | ± | 0.57 |
| Kaempferol | 4.19 | ± | 0.27 |
| Caffeic acid phenethyl ester | 0.35 | ± | 0.01 |
| Apigenin | 0.19 | ± | 0.10 |
| Artepillin C | 0.05 | ± | 0.04 |
| p-Coumaric acid | 0.03 | ± | 0.01 |
| Caffeic acid | 0.01 | ± | 0.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, T.L.; Lee, C.-H.; Han, N.; Back, H.-M.; Rhee, S.-J.; Noh, K.; Yun, H.-Y.; Kang, W.; Chae, J.-W. Application of an Inter-Species Extrapolation Method for the Prediction of Drug Interactions between Propolis and Duloxetine in Humans. Int. J. Mol. Sci. 2020, 21, 1862. https://doi.org/10.3390/ijms21051862
Ngo TL, Lee C-H, Han N, Back H-M, Rhee S-J, Noh K, Yun H-Y, Kang W, Chae J-W. Application of an Inter-Species Extrapolation Method for the Prediction of Drug Interactions between Propolis and Duloxetine in Humans. International Journal of Molecular Sciences. 2020; 21(5):1862. https://doi.org/10.3390/ijms21051862
Chicago/Turabian StyleNgo, Thi Lien, Chung-Hee Lee, Nayoung Han, Hyun-Moon Back, Su-Jin Rhee, Keumhan Noh, Hwi-Yeol Yun, Wonku Kang, and Jung-Woo Chae. 2020. "Application of an Inter-Species Extrapolation Method for the Prediction of Drug Interactions between Propolis and Duloxetine in Humans" International Journal of Molecular Sciences 21, no. 5: 1862. https://doi.org/10.3390/ijms21051862
APA StyleNgo, T. L., Lee, C. -H., Han, N., Back, H. -M., Rhee, S. -J., Noh, K., Yun, H. -Y., Kang, W., & Chae, J. -W. (2020). Application of an Inter-Species Extrapolation Method for the Prediction of Drug Interactions between Propolis and Duloxetine in Humans. International Journal of Molecular Sciences, 21(5), 1862. https://doi.org/10.3390/ijms21051862