New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells

 ,
,  , , ,
, , ,  ,
,  ,
,  ,
,  and
and 
Abstract
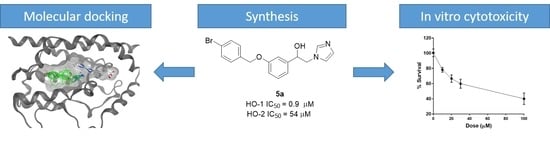
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. HO Inhibition and Structure–Activity Relationships (SARs)
2.3. Docking Studies
2.4. In Vitro Cytotoxic Activity
3. Materials and Methods
3.1. Chemistry
Synthesis of 1-bromo-4-((4-(2-bromoethyl)phenoxy)methyl)benzene (1b)
3.2. General Procedure for the Synthesis of Phenylethylimidazole Derivatives (2a,b)
3.2.1. 1-(3-bromophenethyl)-1H-imidazole (2a)
3.2.2. 1-(4-((4-bromobenzyl)oxy)phenethyl)-1H-imidazole (2b)
3.2.3. Synthesis of 1-(2-((1,1’-biphenyl)-3-yl)ethyl)-1H-imidazole (2c)
3.3. General Procedure for the Synthesis of 1-substituted-2-(1H-imidazol-1-yl)ethanones (4a–f)
3.3.1. 1-[3-[(4-bromobenzyl)oxy]phenyl]2-(1H-imidazol-1-yl)ethanone (4a)
3.3.2. 1-[2-[(4-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanone (4b)
3.3.3. 1-[4-[(3-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanone (4c)
3.3.4. 1-[4-[(2-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanone (4d)
3.3.5. 1-[3-[(3-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanone (4e)
3.3.6. 1-[3-[(2-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanone (4f)
3.4. General Procedure for the Synthesis of 1-(substituted)-2-(1H-imidazol-1-yl)ethanoles 5a–f
3.4.1. 1-[3-[(4-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanol (5a)
3.4.2. 1-[2-[(4-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanol (5b)
3.4.3. 1-[4-[(3-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanol (5c)
3.4.4. 1-[4-[(2-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanol (5d)
3.4.5. 1-[3-[(3-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanol (5e)
3.4.6. 1-[3-[(2-bromobenzyl)oxy]phenyl]-2-(1H-imidazol-1-yl)ethanol (5f)
3.5. General Procedure for the Synthesis of Benzyl Derivatives (6a,b)
3.5.1. 1-(2-(3-bromophenyl)-2-((4-chlorobenzyl)oxy)ethyl)-1H-imidazole (6a)
3.5.2. 1-(2-(3-bromophenyl)-2-((2,5-dichlorobenzyl)oxy)ethyl)-1H-imidazole (6b)
3.6. Biological Evaluation
3.6.1. Preparation of Spleen and Brain Microsomal Fractions
3.6.2. Preparation of Biliverdin Reductase
3.6.3. Measurement of HO-1 and HO-2 Enzymatic Activities in the Microsomal Fraction of Rat Spleen and Brain
3.6.4. Cell Cultures
3.6.5. In Vitro Cytotoxic Activity
3.7. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ogun, A.S.; Valentine, M. Biochemistry, Heme Synthesis; StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandiz, M.L.; Devesa, I. Inducers of heme oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.L.; Johns, D.G.; Kriesgfeld, L.J.; Klein, S.L.; Calvin, D.C.; Demas, G.E.; Schramm, L.P.; Tonegawa, S.; Nelson, R.J.; Snyder, S.H.; et al. Ejaculatory abnormalities in mice with targeted disruption of the gene for heme oxygenase-2. Nat. Med. 1998, 4, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Ragsdale, S.W. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J. Biol. Chem. 2007, 282, 21056–21067. [Google Scholar] [CrossRef] [Green Version]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Foresti, R.; Green, C.J.; Motterlini, R. Generation of bile pigments by haem oxygenase: A refined cellular strategy in response to stressful insults. Biochem. Soc. Symp. 2004, 177–192. [Google Scholar] [CrossRef]
- Drummond, H.A.; Mitchell, Z.L.; Abraham, N.G.; Stec, D.E. Targeting Heme Oxygenase-1 in Cardiovascular and Kidney Disease. Antioxidants 2019, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Sorrenti, V.; Raffaele, M.; Vanella, L.; Acquaviva, R.; Salerno, L.; Pittalà, V.; Intagliata, S.; Di Giacomo, C. Protective Effects of Caffeic Acid Phenethyl Ester (CAPE) and Novel Cape Analogue as Inducers of Heme Oxygenase-1 in Streptozotocin-Induced Type 1 Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 2441. [Google Scholar] [CrossRef] [Green Version]
- Pittalà, V.; Vanella, L.; Salerno, L.; Romeo, G.; Marrazzo, A.; Di Giacomo, C.; Sorrenti, V. Effects of Polyphenolic Derivatives on Heme Oxygenase-System in Metabolic Dysfunctions. Curr. Med. Chem. 2018, 25, 1577–1595. [Google Scholar] [CrossRef]
- Carota, G.; Raffaele, M.; Sorrenti, V.; Salerno, L.; Pittalà, V.; Intagliata, S. Ginseng and heme oxygenase-1: The link between an old herb and a new protective system. Fitoterapia 2019, 139, 104370. [Google Scholar] [CrossRef]
- Son, Y.; Lee, J.H.; Chung, H.T.; Pae, H.O. Therapeutic roles of heme oxygenase-1 in metabolic diseases: Curcumin and resveratrol analogues as possible inducers of heme oxygenase-1. Oxidative Med. Cell. Longev. 2013, 2013, 639541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittalà, V.; Vanella, L.; Maria Platania, C.B.; Salerno, L.; Raffaele, M.; Amata, E.; Marrazzo, A.; Floresta, G.; Romeo, G.; Greish, K.; et al. Synthesis, in vitro and in silico studies of HO-1 inducers and lung antifibrotic agents. Future Med. Chem. 2019, 11, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Pittalà, V.; Vanella, L.; Salerno, L.; Di Giacomo, C.; Acquaviva, R.; Raffaele, M.; Romeo, G.; Modica, M.N.; Prezzavento, O.; Sorrenti, V. Novel Caffeic Acid Phenethyl Ester (Cape) Analogues as Inducers of Heme Oxygenase-1. Curr. Pharm. Des. 2017, 23, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Podkalicka, P.; Mucha, O.; Jozkowicz, A.; Dulak, J.; Loboda, A. Heme oxygenase inhibition in cancers: Possible tools and targets. Contemp. Oncol. 2018, 22, 23–32. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Han, L.; Jiang, J.; Ma, Q.; Wu, Z.; Wang, Z. The inhibition of heme oxygenase-1 enhances the chemosensitivity and suppresses the proliferation of pancreatic cancer cells through the SHH signaling pathway. Int. J. Oncol. 2018, 52, 2101–2109. [Google Scholar] [CrossRef]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrenti, V.; Pittalà, V.; Romeo, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Turnaturi, R.; Prezzavento, O.; Barbagallo, I.; Vanella, L.; et al. Targeting heme Oxygenase-1 with hybrid compounds to overcome Imatinib resistance in chronic myeloid leukemia cell lines. Eur. J. Med. Chem. 2018, 158, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Romeo, G.; Modica, M.N.; Amata, E.; Sorrenti, V.; Barbagallo, I.; Pittalà, V. Heme oxygenase-1: A new druggable target in the management of chronic and acute myeloid leukemia. Eur. J. Med. Chem. 2017, 142, 163–178. [Google Scholar] [CrossRef]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Marrazzo, A.; Siracusa, M.A.; Sorrenti, V.; Di Giacomo, C.; Vanella, L.; Parayath, N.N.; et al. Novel imidazole derivatives as heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) inhibitors and their cytotoxic activity in human-derived cancer cell lines. Eur. J. Med. Chem. 2015, 96, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Mucha, O.; Podkalicka, P.; Mikulski, M.; Barwacz, S.; Andrysiak, K.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Ryszawy, D.; Bialas, A.; et al. Development and characterization of a new inhibitor of heme oxygenase activity for cancer treatment. Arch. Biochem. Biophys. 2019, 671, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbagallo, I.; Giallongo, C.; Volti, G.L.; Distefano, A.; Camiolo, G.; Raffaele, M.; Salerno, L.; Pittalà, V.; Sorrenti, V.; Avola, R.; et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol. Neurobiol. 2018, 56, 1451–1460. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vukomanovic, D.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. Structural insights into human heme oxygenase-1 inhibition by potent and selective azole-based compounds. J. R. Soc. Interface 2013, 10, 20120697. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vukomanovic, D.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. Structural Insights into Azole-based Inhibitors of Heme Oxygenase-1: Development of Selective Compounds for Therapeutic Applications. Curr. Med. Chem. 2018, 25, 5803–5821. [Google Scholar] [CrossRef]
- Salerno, L.; Floresta, G.; Ciaffaglione, V.; Gentile, D.; Margani, F.; Turnaturi, R.; Rescifina, A.; Pittalà, V. Progress in the development of selective heme oxygenase-1 inhibitors and their potential therapeutic application. Eur. J. Med. Chem. 2019, 167, 439–453. [Google Scholar] [CrossRef]
- Intagliata, S.; Salerno, L.; Ciaffaglione, V.; Leonardi, C.; Fallica, A.N.; Carota, G.; Amata, E.; Marrazzo, A.; Pittalà, V.; Romeo, G. Heme Oxygenase-2 (HO-2) as a therapeutic target: Activators and inhibitors. Eur. J. Med. Chem. 2019, 183, 111703. [Google Scholar] [CrossRef]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Siracusa, M.A.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Tibullo, D.; Sorrenti, V. Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg. Med. Chem. 2013, 21, 5145–5153. [Google Scholar] [CrossRef]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittalà, V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Comprehensive data on a 2D-QSAR model for Heme Oxygenase isoform 1 inhibitors. Data Br. 2017, 15, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Heme Oxygenase Database (HemeOxDB) and QSAR Analysis of Isoform 1 Inhibitors. ChemMedChem 2017, 12, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-M.; Li, X.-M.; Xu, W.; Li, F.; Wang, J.; Kong, L.-Y.; Wang, X.-B. Acetophenone derivatives: Novel and potent small molecule inhibitors of monoamine oxidase B. MedChemComm 2015, 6, 2146–2157. [Google Scholar] [CrossRef]
- Roman, G.; Mares, M.; Nastasa, V. A novel antifungal agent with broad spectrum: 1-(4-biphenylyl)-3-(1H-imidazol-1-yl)-1-propanone. Arch. Pharm. 2013, 346, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Geng, M.; Teng, P.; Zhao, D.; Lu, X.; Li, J.X. Ultrasound-promoted intramolecular direct arylation in a capillary flow microreactor. Ultrason. Sonochem. 2012, 19, 250–256. [Google Scholar] [CrossRef]
- Rahman, M.N.; Vlahakis, J.Z.; Vukomanovic, D.; Lee, W.; Szarek, W.A.; Nakatsu, K.; Jia, Z. A novel, “double-clamp” binding mode for human heme oxygenase-1 inhibition. PLoS ONE 2012, 7, e29514. [Google Scholar] [CrossRef] [Green Version]
- Sugishima, M.; Higashimoto, Y.; Oishi, T.; Takahashi, H.; Sakamoto, H.; Noguchi, M.; Fukuyama, K. X-ray crystallographic and biochemical characterization of the inhibitory action of an imidazole-dioxolane compound on heme oxygenase. Biochemistry 2007, 46, 1860–1867. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Kinobe, R.T.; Bowers, R.J.; Brien, J.F.; Nakatsu, K.; Szarek, W.A. Synthesis and evaluation of azalanstat analogues as heme oxygenase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1457–1461. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Csizmadia, F. JChem: Java applets and modules supporting chemical database handling from web browsers. J. Chem. Inf. Comput. Sci. 2000, 40, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–105. [Google Scholar] [CrossRef]
- Galimberti, M.; Barbera, V.; Guerra, S.; Bernardi, A. Facile Functionalization of Sp(2) Carbon Allotropes with a Biobased Janus Molecule. Rubber Chem. Technol. 2017, 90, 285–307. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | X | IC50 (μM) ± SD a | |
|---|---|---|---|---|
| HO-1 | HO-2 | |||
| 2a | 3-Br | CH2 | 100 ± 5.60 | NT |
| 2b | 4-(4-BrC6H4)CH2O | CH2 | 62.87 ± 3.20 | NT |
| 2c | 3-Ph | CH2 | 46.77 ± 1.80 | NT |
| 4gb | 3-Br | CO | 38.17 ± 1.80 | NT |
| 4hb | 3-Ph | CO | 19.87 ± 2.50 | NT |
| 4ib | 4-(4-BrC6H4)CH2O | CO | 55.46 ± 0.05 | NT |
| 5a | 3-(4-BrC6H4)CH2O | CHOH | 0.90 ± 0.02 | 53.59 ±1.20 |
| 5b | 2-(4-BrC6H4)CH2O | CHOH | >100 | NT |
| 5c | 4-(3-BrC6H4)CH2O | CHOH | 41 ± 1.50 | NT |
| 5d | 4-(2-BrC6H4)CH2O | CHOH | 9 ± 2.20 | 15.85 ±1.60 |
| 5e | 3-(3-BrC6H4)CH2O | CHOH | 46 ± 1.90 | NT |
| 5f | 3-(2-BrC6H4)CH2O | CHOH | 44 ± 1.80 | NT |
| 6a | 3-Br | CHOCH2(4-ClC6H4) | >100 | NT |
| 6b | 3-Br | CHOCH2(2,5-ClC6H3) | 66 ± 3.20 | NT |
| A | 3-Br | CHOH | 0.40 ± 0.01 c | 32 ± 2.2 c |
| B | 3-Ph | CHOH | 0.90 ± 0.08 c | >100 c |
| C | 4-(4-BrC6H4)CH2O | CHOH | 0.95 ± 0.02 c | >100 c |
| SnPP | - | - | 0.58 ± 0.03 | 0.36 ± 0.01 |
| Compound | ΔGB calcd. (kcal/mol) | Ki calcd. (μM) | IC50 exp. (μM) HO-1 |
|---|---|---|---|
| 2a | −5.71 | 64.92 | 100 |
| 2b | −5.91 | 46.31 | 62.87 ± 3.20 |
| 2c | −6.10 | 33.60 | 46.77 ± 1.80 |
| 5a | −8.42 | 0.66 | 0.9 ± 0.02 |
| 5b | −5.48 | 95.74 | >100 |
| 5c | −5.38 | 113.35 | 41 ± 1.50 |
| 5d | −6.60 | 14.44 | 9 ± 2.20 |
| 5e | −6.15 | 30.88 | 46 ± 1.90 |
| 5f | −6.15 | 30.88 | 44 ± 1.80 |
| 6a | −4.24 | 777.11 | >100 |
| 6b | −5.81 | 54.83 | 66 ± 3.20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciaffaglione, V.; Intagliata, S.; Pittalà, V.; Marrazzo, A.; Sorrenti, V.; Vanella, L.; Rescifina, A.; Floresta, G.; Sultan, A.; Greish, K.; et al. New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 1923. https://doi.org/10.3390/ijms21061923
Ciaffaglione V, Intagliata S, Pittalà V, Marrazzo A, Sorrenti V, Vanella L, Rescifina A, Floresta G, Sultan A, Greish K, et al. New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(6):1923. https://doi.org/10.3390/ijms21061923
Chicago/Turabian StyleCiaffaglione, Valeria, Sebastiano Intagliata, Valeria Pittalà, Agostino Marrazzo, Valeria Sorrenti, Luca Vanella, Antonio Rescifina, Giuseppe Floresta, Ameera Sultan, Khaled Greish, and et al. 2020. "New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 6: 1923. https://doi.org/10.3390/ijms21061923
APA StyleCiaffaglione, V., Intagliata, S., Pittalà, V., Marrazzo, A., Sorrenti, V., Vanella, L., Rescifina, A., Floresta, G., Sultan, A., Greish, K., & Salerno, L. (2020). New Arylethanolimidazole Derivatives as HO-1 Inhibitors with Cytotoxicity against MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences, 21(6), 1923. https://doi.org/10.3390/ijms21061923