The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer

Abstract
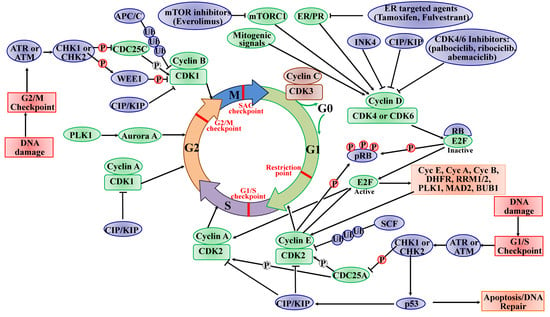
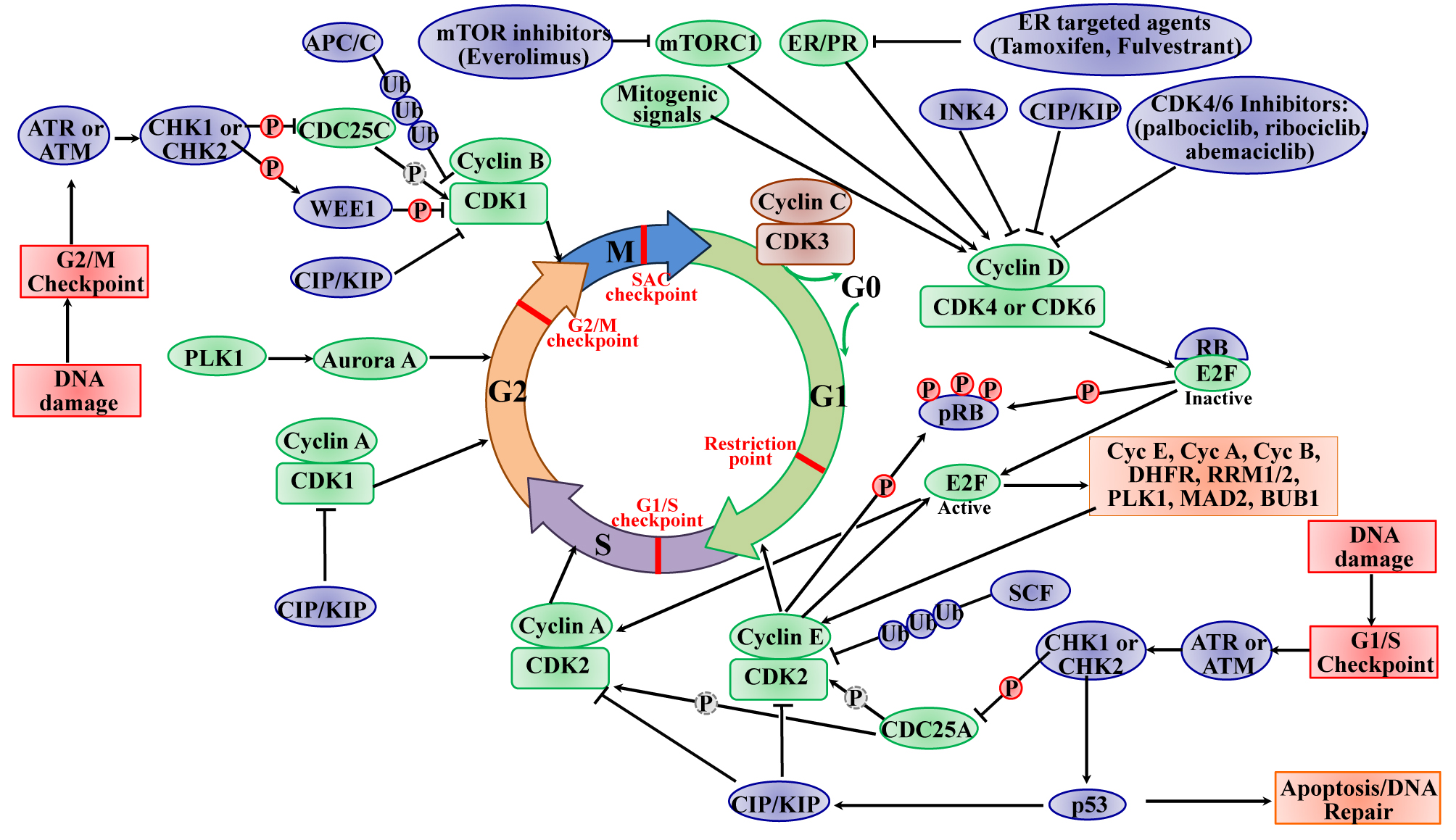
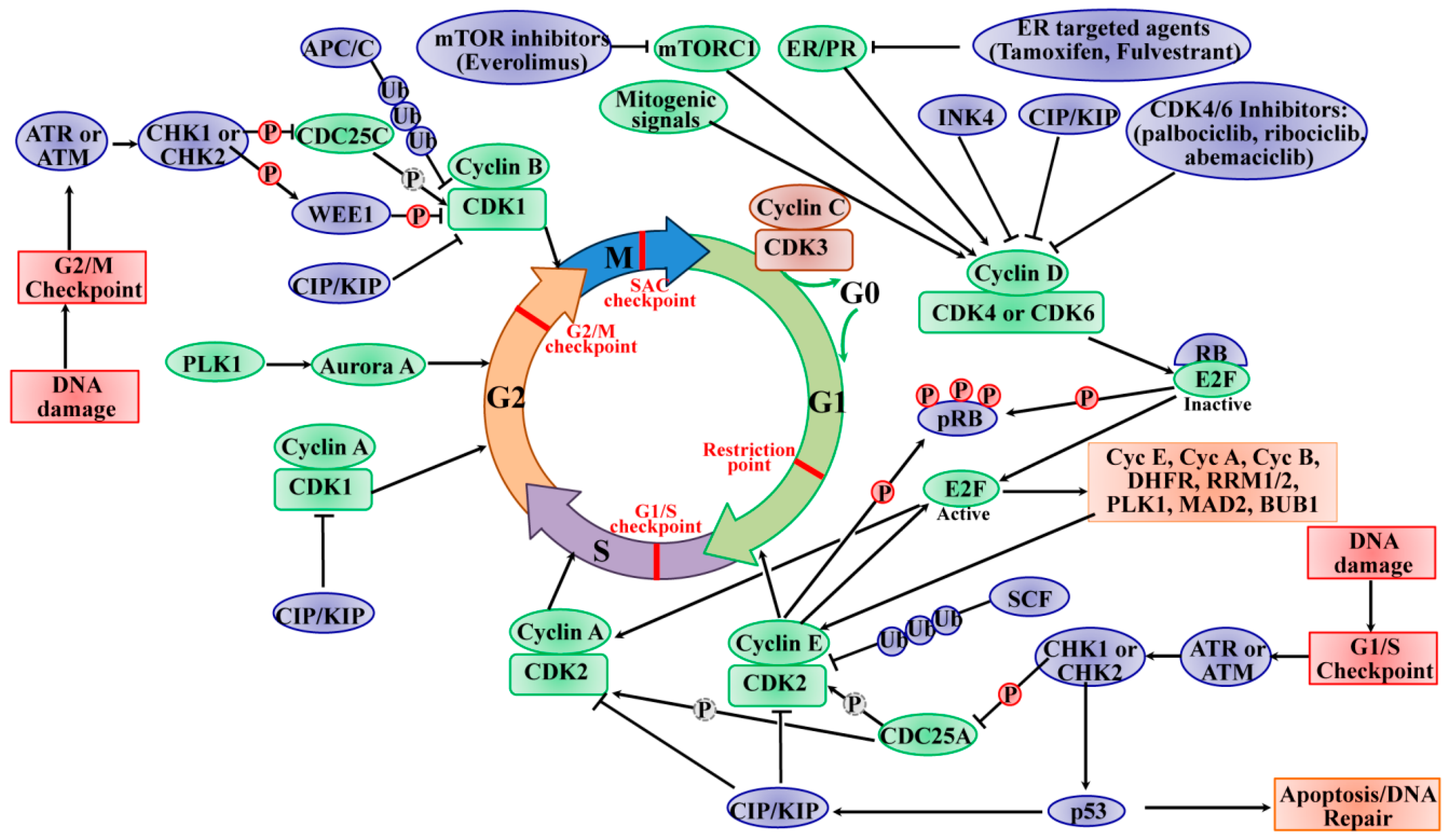
:
1. Introduction
2. CDKs in the Cell Cycle and Transcription
2.1. The Roles of CDKs in the Cell Cycle
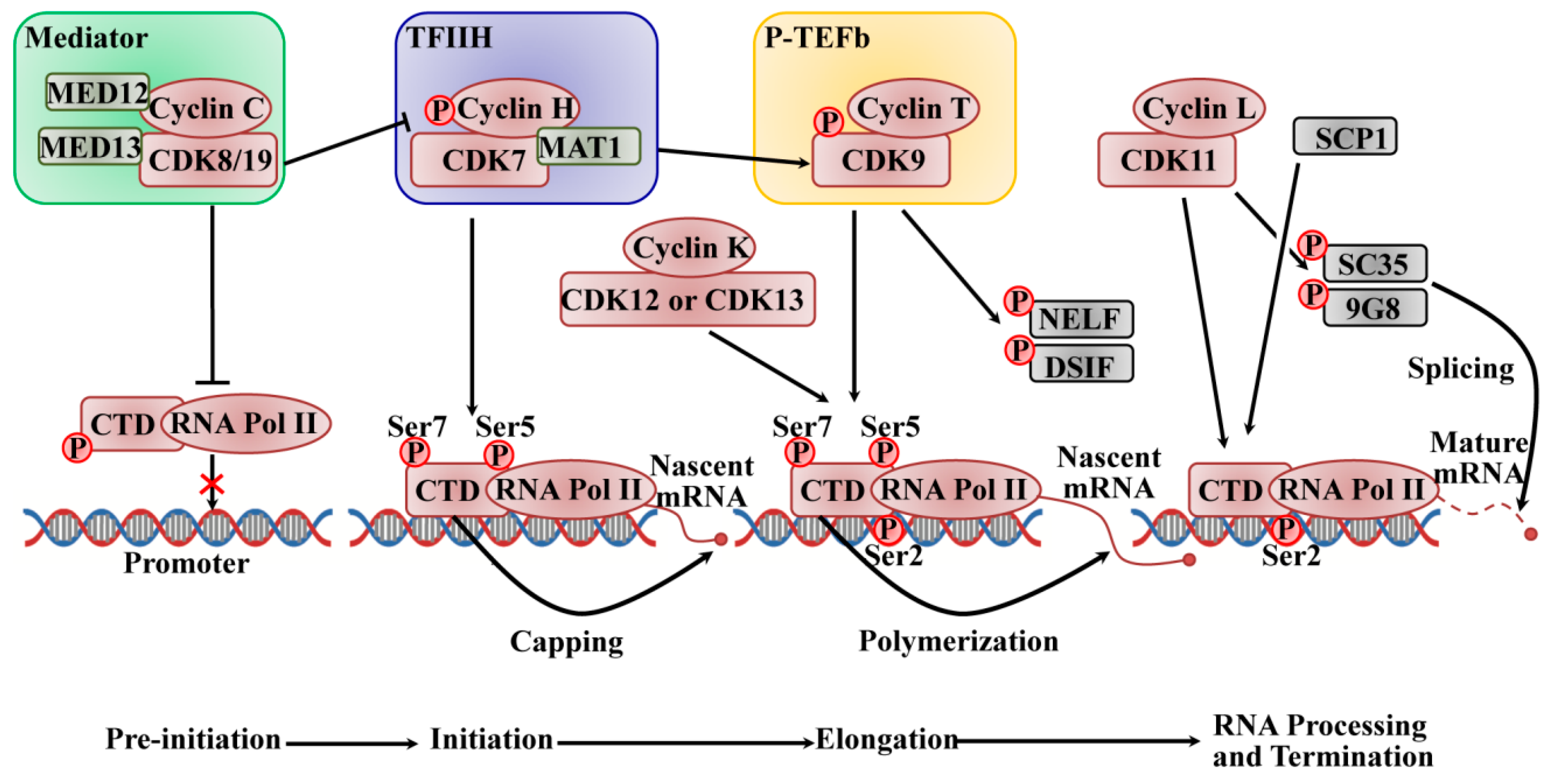
2.2. The Roles of CDKs in Transcription
3. Dysregulation of CDKs in BC
4. Targeting CDKs in BC Therapy
4.1. The Early Pan-CDK Inhibitors in BC
4.2. The Clinical Success of CDK4/6-Selective Inhibitors in BC
4.3. The Novel CDK Inhibitors in BC
4.4. The Combined Treatment with CDK Inhibitors and Other Agents
5. Conclusions
Authors Contributions
Funding
Conflicts of Interest
References
- Ding, L.; Gu, H.; Xiong, X.; Ao, H.; Cao, J.; Lin, W.; Yu, M.; Lin, J.; Cui, Q. MicroRNAs Involved in Carcinogenesis, Prognosis, Therapeutic Resistance and Applications in Human Triple-Negative Breast Cancer. Cells 2019, 8, 1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxmen, A. The hard facts. Nature 2012, 485, S50–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathe, A.; Scott, R.J.; Avery-Kiejda, K.A. MiRNAs and Other Epigenetic Changes as Biomarkers in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2015, 16, 28347–28376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Thu, K.L.; Soria-Bretones, I.; Mak, T.W.; Cescon, D.W. Targeting the cell cycle in breast cancer: Towards the next phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Xu, J.; Sun, T. Cyclin-Dependent Kinases 4/6 Inhibitors in Breast Cancer: Current Status, Resistance, and Combination Strategies. J. Cancer 2019, 10, 5504–5517. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; He, M.; Shah, A.A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015-2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Canavese, M.; Santo, L.; Raje, N. Cyclin dependent kinases in cancer: Potential for therapeutic intervention. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol. Biol. 2014, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Valius, M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Ma, T.; Van Tine, B.A.; Wei, Y.; Garrett, M.D.; Nelson, D.; Adams, P.D.; Wang, J.; Qin, J.; Chow, L.T.; Harper, J.W. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000, 14, 2298–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, M.; Horn, H.F.; Tarapore, P.; Tokuyama, Y.; Smulian, A.G.; Chan, P.K.; Knudsen, E.S.; Hofmann, I.A.; Snyder, J.D.; Bove, K.E.; et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 2000, 103, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Sever-Chroneos, Z.; Angus, S.P.; Fribourg, A.F.; Wan, H.; Todorov, I.; Knudsen, K.E.; Knudsen, E.S. Retinoblastoma tumor suppressor protein signals through inhibition of cyclin-dependent kinase 2 activity to disrupt PCNA function in S phase. Mol. Cell Biol. 2001, 21, 4032–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Rollins, B.J. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [Green Version]
- Santo, L.; Siu, K.T.; Raje, N. Targeting Cyclin-Dependent Kinases and Cell Cycle Progression in Human Cancers. Semin. Oncol. 2015, 42, 788–800. [Google Scholar] [CrossRef]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef] [Green Version]
- Koniaras, K.; Cuddihy, A.R.; Christopoulos, H.; Hogg, A.; O’Connell, M.J. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 2001, 20, 7453–7463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, S.L.; Charlet, A.; Haas, W.; Gygi, S.P.; Kellogg, D.R. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 2005, 122, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The spindle assembly checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Suh, H.; Hazelbaker, D.Z.; Soares, L.M.; Buratowski, S. The C-terminal domain of Rpb1 functions on other RNA polymerase II subunits. Mol. Cell 2013, 51, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Jeronimo, C.; Bataille, A.R.; Robert, F. The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem. Rev. 2013, 113, 8491–8522. [Google Scholar] [CrossRef]
- Jeronimo, C.; Collin, P.; Robert, F. The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J. Mol. Biol. 2016, 428, 2607–2622. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 10, 118–136. [Google Scholar] [CrossRef]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Yin, J.W.; Wang, G. The Mediator complex: A master coordinator of transcription and cell lineage development. Development 2014, 141, 977–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2012, 3, 731–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Viladevall, L.; St Amour, C.V.; Rosebrock, A.; Schneider, S.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Schwer, B.; Leatherwood, J.K.; Fisher, R.P. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol. Cell 2009, 33, 738–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Blanco, A.; Sen, N.; Mayan-Santos, M.; Sacristan, M.P.; Graham, B.; Jarmuz, A.; Giess, A.; Webb, E.; Game, L.; Eick, D.; et al. Cdc14 phosphatase promotes segregation of telomeres through repression of RNA polymerase II transcription. Nat. Cell. Biol. 2011, 13, 1450–1456. [Google Scholar] [CrossRef] [Green Version]
- Guillamot, M.; Manchado, E.; Chiesa, M.; Gomez-Lopez, G.; Pisano, D.G.; Sacristan, M.P.; Malumbres, M. Cdc14b regulates mammalian RNA polymerase II and represses cell cycle transcription. Sci. Rep. 2011, 1, 189. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Dominguez-Brauer, C.; Wang, Z.; Asara, J.M.; Costa, R.H.; Tyner, A.L.; Lau, L.F.; Raychaudhuri, P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J. Biol. Chem. 2009, 284, 30695–30707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marais, A.; Ji, Z.; Child, E.S.; Krause, E.; Mann, D.J.; Sharrocks, A.D. Cell cycle-dependent regulation of the forkhead transcription factor FOXK2 by CDK.cyclin complexes. J. Biol. Chem. 2010, 285, 35728–35739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Wei, Y.; Zhang, F.; Hsu, Y.H.; Chan, L.C.; Xia, W.; Ke, B.; Zhu, C.; Deng, R.; Tang, J.; et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat. Commun. 2019, 10, 5114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Y.; Wang, C.; Ju, H.; Liu, W.; Zhang, X.; Miao, S.; Wang, L.; Sun, Q.; Song, W. Rhomboid domain-containing protein 1 promotes breast cancer progression by regulating the p-Akt and CDK2 levels. Cell Commun. Signal. 2018, 16, 65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Huang, A.; Zhang, A.; Zhou, C. HuR promotes breast cancer cell proliferation and survival via binding to CDK3 mRNA. Biomed. Pharmacother 2017, 91, 788–795. [Google Scholar] [CrossRef]
- Cao, T.; Xiao, T.; Huang, G.; Xu, Y.; Zhu, J.J.; Wang, K.; Ye, W.; Guan, H.; He, J.; Zheng, D. CDK3, target of miR-4469, suppresses breast cancer metastasis via inhibiting Wnt/beta-catenin pathway. Oncotarget 2017, 8, 84917–84927. [Google Scholar]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef] [Green Version]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. Loss of Cdk5 in breast cancer cells promotes ROS-mediated cell death through dysregulation of the mitochondrial permeability transition pore. Oncogene 2018, 37, 1788–1804. [Google Scholar] [CrossRef] [Green Version]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. Targeting Cdk5 for killing of breast cancer cells via perturbation of redox homeostasis. Oncoscience 2018, 5, 152–154. [Google Scholar]
- Liang, Q.; Li, L.; Zhang, J.; Lei, Y.; Wang, L.; Liu, D.X.; Feng, J.; Hou, P.; Yao, R.; Zhang, Y.; et al. CDK5 is essential for TGF-beta1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013, 3, 2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ni Chonghaile, T.; Fan, Y.; Madden, S.F.; Klinger, R.; O’Connor, A.E.; Walsh, L.; O’Hurley, G.; Mallya Udupi, G.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemet, J.; Jelicic, B.; Rubelj, I.; Sopta, M. The two faces of Cdk8, a positive/negative regulator of transcription. Biochimie 2014, 97, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Crown, J. CDK8: A new breast cancer target. Oncotarget 2017, 8, 14269–14270. [Google Scholar] [CrossRef]
- Broude, E.V.; Gyorffy, B.; Chumanevich, A.A.; Chen, M.; McDermott, M.S.; Shtutman, M.; Catroppo, J.F.; Roninson, I.B. Expression of CDK8 and CDK8-interacting Genes as Potential Biomarkers in Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Schlafstein, A.J.; Withers, A.E.; Rudra, S.; Danelia, D.; Switchenko, J.M.; Mister, D.; Harari, S.; Zhang, H.; Daddacha, W.; Ehdaivand, S.; et al. CDK9 Expression Shows Role as a Potential Prognostic Biomarker in Breast Cancer Patients Who Fail to Achieve Pathologic Complete Response after Neoadjuvant Chemotherapy. Int. J. Breast Cancer 2018. [Google Scholar] [CrossRef] [Green Version]
- Del Re, M.; Bertolini, I.; Crucitta, S.; Fontanelli, L.; Rofi, E.; De Angelis, C.; Diodati, L.; Cavallero, D.; Gianfilippo, G.; Salvadori, B.; et al. Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Res. Treat. 2019, 178, 57–62. [Google Scholar] [CrossRef]
- Guen, V.J.; Gamble, C.; Lees, J.A.; Colas, P. The awakening of the CDK10/Cyclin M protein kinase. Oncotarget 2017, 8, 50174–50186. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Li, H.; Qin, X.; Zhang, Y.; Song, W.; Ran, Y.; Gao, F. Decreased CDK10 expression correlates with lymph node metastasis and predicts poor outcome in breast cancer patients - a short report. Cell Oncol. (Dordr) 2015, 38, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Turner, N.C.; Elliott, R.; Syed, N.; Garrone, O.; Gasco, M.; Tutt, A.N.; Crook, T.; Lord, C.J.; Ashworth, A. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell 2008, 13, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos Paparidis, N.F.; Canduri, F. The Emerging Picture of CDK11: Genetic, Functional and Medicinal Aspects. Curr. Med. Chem. 2018, 25, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Loyer, P.; Trembley, J.H.; Grenet, J.A.; Busson, A.; Corlu, A.; Zhao, W.; Kocak, M.; Kidd, V.J.; Lahti, J.M. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: Influence of cyclin L isoforms on splice site selection. J. Biol. Chem. 2008, 283, 7721–7732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Y.; Huang, S.; Peng, H.; Liu, M.; Zhao, J.; Shao, Z.; Wu, J. Critical role of CDK11(p58) in human breast cancer growth and angiogenesis. BMC Cancer 2015, 15, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Han, C.; Li, D.; Yu, Z.; Li, F.; Li, F.; An, Q.; Bai, H.; Zhang, X.; Duan, Z.; et al. Cyclin-dependent kinase 11(p110) (CDK11(p110)) is crucial for human breast cancer cell proliferation and growth. Sci. Rep. 2015, 5, 10433. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shen, J.K.; Hornicek, F.J.; Kan, Q.; Duan, Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget 2016, 7, 40846–40859. [Google Scholar] [CrossRef] [Green Version]
- Tien, J.F.; Mazloomian, A.; Cheng, S.G.; Hughes, C.S.; Chow, C.C.T.; Canapi, L.T.; Oloumi, A.; Trigo-Gonzalez, G.; Bashashati, A.; Xu, J.; et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017, 45, 6698–6716. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, K.; Wai, P.T.; Maguire, S.L.; Daley, F.; Haider, S.; Kriplani, D.; Campbell, J.; Mirza, H.; Grigoriadis, A.; Tutt, A.; et al. Evaluation of CDK12 Protein Expression as a Potential Novel Biomarker for DNA Damage Response-Targeted Therapies in Breast Cancer. Mol. Cancer Ther. 2018, 17, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558. [Google Scholar] [CrossRef]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohoutek, J.; Blazek, D. Cyclin K goes with Cdk12 and Cdk13. Cell Div. 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, G.; Niehrs, C. Emerging links between CDK cell cycle regulators and Wnt signaling. Trends Cell Biol. 2010, 20, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, J.; Huang, L.H.; Sun, X.X. Cyclin-Dependent Kinase 14 Promotes Cell Proliferation, Migration and Invasion in Ovarian Cancer by Inhibiting Wnt Signaling Pathway. Gynecol Obstet Invest. 2017, 82, 230–239. [Google Scholar] [CrossRef]
- Wang, B.; Zou, A.; Ma, L.; Chen, X.; Wang, L.; Zeng, X.; Tan, T. miR-455 inhibits breast cancer cell proliferation through targeting CDK14. Eur. J. Pharmacol. 2017, 807, 138–143. [Google Scholar] [CrossRef]
- Imawari, Y.; Mimoto, R.; Hirooka, S.; Morikawa, T.; Takeyama, H.; Yoshida, K. Downregulation of dual-specificity tyrosine-regulated kinase 2 promotes tumor cell proliferation and invasion by enhancing cyclin-dependent kinase 14 expression in breast cancer. Cancer Sci. 2018, 109, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Fujimoto, A.; Furuta, M.; Tanaka, H.; Chiba, K.; Boroevich, K.A.; Abe, T.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS ONE 2014, 9, e114263. [Google Scholar] [CrossRef]
- Li, S.; Dai, X.; Gong, K.; Song, K.; Tai, F.; Shi, J. PA28alpha/beta Promote Breast Cancer Cell Invasion and Metastasis via Down-Regulation of CDK15. Front. Oncol. 2019, 9, 1283. [Google Scholar] [CrossRef] [Green Version]
- Yanagi, T.; Matsuzawa, S. PCTAIRE1/PCTK1/CDK16: A new oncotarget? Cell Cycle 2015, 14, 463–464. [Google Scholar] [CrossRef] [Green Version]
- Ou, C.Y.; Poon, V.Y.; Maeder, C.I.; Watanabe, S.; Lehrman, E.K.; Fu, A.K.; Park, M.; Fu, W.Y.; Jorgensen, E.M.; Ip, N.Y.; et al. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 2010, 141, 846–858. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Watanabe, S.; Poon, V.Y.; Ou, C.Y.; Jorgensen, E.M.; Shen, K. CYY-1/cyclin Y and CDK-5 differentially regulate synapse elimination and formation for rewiring neural circuits. Neuron. 2011, 70, 742–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagi, T.; Shi, R.; Aza-Blanc, P.; Reed, J.C.; Matsuzawa, S. PCTAIRE1-knockdown sensitizes cancer cells to TNF family cytokines. PLoS ONE 2015, 10, e0119404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaput, D.; Kirouac, L.; Stevens, S.M., Jr.; Padmanabhan, J. Potential role of PCTAIRE-2, PCTAIRE-3 and P-Histone H4 in amyloid precursor protein-dependent Alzheimer pathology. Oncotarget 2016, 7, 8481–8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, M.; Perna, E.; Tronnolone, S.; Colecchia, D.; Chiariello, M. Activated kinase screening identifies the IKBKE oncogene as a positive regulator of autophagy. Autophagy 2019, 15, 312–326. [Google Scholar] [CrossRef] [Green Version]
- Valladares, A.; Hernandez, N.G.; Gomez, F.S.; Curiel-Quezada, E.; Madrigal-Bujaidar, E.; Vergara, M.D.; Martinez, M.S.; Arenas Aranda, D.J. Genetic expression profiles and chromosomal alterations in sporadic breast cancer in Mexican women. Cancer Genet. Cytogenet 2006, 170, 147–151. [Google Scholar] [CrossRef]
- Barone, G.; Staples, C.J.; Ganesh, A.; Patterson, K.W.; Bryne, D.P.; Myers, K.N.; Patil, A.A.; Eyers, C.E.; Maslen, S.; Skehel, J.M.; et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 2016, 44, 8772–8785. [Google Scholar] [CrossRef] [Green Version]
- Braams, E.; D’Angiolella, V. Keeping CDK18 in balance to prevent DNA replication stress in breast cancer. Oncotarget 2018, 9, 37610–37611. [Google Scholar] [CrossRef]
- Barone, G.; Arora, A.; Ganesh, A.; Abdel-Fatah, T.; Moseley, P.; Ali, R.; Chan, S.Y.; Savva, C.; Schiavone, K.; Carmell, N.; et al. The relationship of CDK18 expression in breast cancer to clinicopathological parameters and therapeutic response. Oncotarget 2018, 9, 29508–29524. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Dong, L.; Zhang, B.; Dai, J.; Zhang, Y.; Wang, Y.; Qin, S. Long noncoding RNA CASC2 promotes paclitaxel resistance in breast cancer through regulation of miR-18a-5p/CDK19. Histochem Cell Biol. 2019, 152, 281–291. [Google Scholar] [CrossRef]
- Dukelow, T.; Kishan, D.; Khasraw, M.; Murphy, C.G. CDK4/6 inhibitors in breast cancer. Anticancer Drugs 2015, 26, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.; Papanikolaou, A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. 2005, 23, 4215–4224. [Google Scholar] [CrossRef] [PubMed]
- Spring, L.M.; Zangardi, M.L.; Moy, B.; Bardia, A. Clinical Management of Potential Toxicities and Drug Interactions Related to Cyclin-Dependent Kinase 4/6 Inhibitors in Breast Cancer: Practical Considerations and Recommendations. Oncologist 2017, 22, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, T.J.; Liu, J.C.; Vizeacoumar, F.; Sun, T.; Maclean, N.; Egan, S.E.; Schimmer, A.D.; Datti, A.; Zacksenhaus, E. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PLoS ONE 2013, 8, e78641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, P.; Sanna, V.; Fancellu, A.; Cinieri, S. A clinical evaluation of treatments that target cell cycle machinery in breast cancer. Expert Opin. Pharmacother 2019, 20, 2305–2315. [Google Scholar] [CrossRef]
- Jabbour-Leung, N.A.; Chen, X.; Bui, T.; Jiang, Y.; Yang, D.; Vijayaraghavan, S.; McArthur, M.J.; Hunt, K.K.; Keyomarsi, K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Van der Noord, V.E.; McLaughlin, R.P.; Smid, M.; Foekens, J.A.; Martens, J.W.M.; Zhang, Y.; van de Water, B. An increased cell cycle gene network determines MEK and Akt inhibitor double resistance in triple-negative breast cancer. Sci. Rep. 2019, 9, 13308. [Google Scholar] [CrossRef] [Green Version]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [Green Version]
- Matutino, A.; Amaro, C.; Verma, S. CDK4/6 inhibitors in breast cancer: Beyond hormone receptor-positive HER2-negative disease. Ther. Adv. Med. Oncol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Aaltonen, K.; Amini, R.M.; Heikkila, P.; Aittomaki, K.; Tamminen, A.; Nevanlinna, H.; Blomqvist, C. High cyclin B1 expression is associated with poor survival in breast cancer. Br. J. Cancer 2009, 100, 1055–1060. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.R.; Walton, M.I.; Garrett, M.D.; Workman, P. The Cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of Cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res. 2004, 64, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, H.; Czech, J.; Naik, R.; Kaur, G.; Worland, P.; Losiewicz, M.; Parker, B.; Carlson, B.; Smith, A.; Senderowicz, A.; et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol. 1996, 9, 1143–1168. [Google Scholar] [CrossRef]
- Rizzolio, F.; Tuccinardi, T.; Caligiuri, I.; Lucchetti, C.; Giordano, A. CDK inhibitors: From the bench to clinical trials. Curr. Drug Targets 2010, 11, 279–290. [Google Scholar] [CrossRef]
- Lin, T.S.; Ruppert, A.S.; Johnson, A.J.; Fischer, B.; Heerema, N.A.; Andritsos, L.A.; Blum, K.A.; Flynn, J.M.; Jones, J.A.; Hu, W.; et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009, 27, 6012–6018. [Google Scholar] [CrossRef]
- Lanasa, M.C.; Andritsos, L.; Brown, J.R.; Gabrilove, J.; Caligaris-Cappio, F.; Ghia, P.; Larson, R.A.; Kipps, T.J.; Leblond, V.; Milligan, D.W.; et al. Final results of EFC6663: A multicenter, international, phase 2 study of alvocidib for patients with fludarabine-refractory chronic lymphocytic leukemia. Leuk. Res. 2015, 39, 495–500. [Google Scholar] [CrossRef] [Green Version]
- Benson, C.; White, J.; De Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.L.; Small, K.; et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Xu, D.; Li, X.; Zhang, J.; Xu, W.; Hou, J.; Zhang, W.; Tang, J. Latest Overview of the Cyclin-Dependent Kinases 4/6 Inhibitors in Breast Cancer: The Past, the Present and the Future. J. Cancer 2019, 10, 6608–6617. [Google Scholar] [CrossRef] [PubMed]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [PubMed]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A.; et al. Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Lorusso, P.M.; Demichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.K.; LoRusso, P.M.; Dickson, M.A.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; Courtney, R.; O’Dwyer, P.J. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br. J. Cancer 2011, 104, 1862–1868. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef] [Green Version]
- Kwapisz, D. Cyclin-dependent kinase 4/6 inhibitors in breast cancer: Palbociclib, ribociclib, and abemaciclib. Breast Cancer Res. Treat. 2017, 166, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Val Bianchi, G.; Esteva, F.J.; Martin, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2019, 382, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet. Oncol. 2016, 17, 425–439. [Google Scholar] [PubMed] [Green Version]
- Turner, N.C.; Ro, J.; Andre, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Sousa, R.; Shapiro, G.I.; Tolaney, S.M. Clinical Development of the CDK4/6 Inhibitors Ribociclib and Abemaciclib in Breast Cancer. Breast Care (Basel) 2016, 11, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S.; et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res. 2016, 22, 5696–5705. [Google Scholar] [CrossRef] [Green Version]
- Curigliano, G.; Gomez Pardo, P.; Meric-Bernstam, F.; Conte, P.; Lolkema, M.P.; Beck, J.T.; Bardia, A.; Martinez Garcia, M.; Penault-Llorca, F.; Dhuria, S.; et al. Ribociclib plus letrozole in early breast cancer: A presurgical, window-of-opportunity study. Breast 2016, 28, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; Andre, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet. Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Spring, L.; Bardia, A. Cycling Toward Progress: Ribociclib, a CDK 4/6 Inhibitor for Breast Cancer. Clin. Cancer Res. 2018, 24, 2981–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Lin, N.U.; Thornton, D.; Klise, S.; Costigan, T.M.; Turner, P.K.; Anders, C.K. Abemaciclib for the treatment of brain metastases (BM) secondary to hormone receptor positive (HR+), HER2 negative breast cancer. J. Clin. Oncol. 2017, 35, 1019. [Google Scholar] [CrossRef]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Spring, L.; Bardia, A.; Modi, S. Targeting the cyclin D-cyclin-dependent kinase (CDK) 4/6-retinoblastoma pathway with selective CDK 4/6 inhibitors in hormone receptor-positive breast cancer: Rationale, current status, and future directions. Discov. Med. 2016, 21, 65–74. [Google Scholar] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ. Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Hurvitz, S.A.; Martin, M.; Press, M.F.; Chan, D.; Fernandez-Abad, M.; Petru, E.; Rostorfer, R.; Guarneri, V.; Huang, C.S.; Barriga, S.; et al. Potent Cell-Cycle Inhibition and Upregulation of Immune Response with Abemaciclib and Anastrozole in neoMONARCH, Phase II Neoadjuvant Study in HR(+)/HER2(-) Breast Cancer. Clin. Cancer Res. 2019, 26, 566–580. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.H.; Ryu, H.; Kim, D.H.; Cheng, W.N.; Yoon, J.E.; Kang, S.; Han, S.G. Piperlongumine Induces Cell Cycle Arrest via Reactive Oxygen Species Accumulation and IKKbeta Suppression in Human Breast Cancer Cells. Antioxidants (Basel) 2019, 8. [Google Scholar]
- Liu, Q.; Cao, Y.; Zhou, P.; Gui, S.; Wu, X.; Xia, Y.; Tu, J. Panduratin A Inhibits Cell Proliferation by Inducing G0/G1 Phase Cell Cycle Arrest and Induces Apoptosis in Breast Cancer Cells. Biomol. Ther. (Seoul) 2018, 26, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Wang, C.Y.; Hu, R.; Lee, J.Y.; Luu, T.T.; Park, H.J.; Lee, S.K. Antitumor Activity of Vanicoside B Isolated from Persicaria dissitiflora by Targeting CDK8 in Triple-Negative Breast Cancer Cells. J. Nat. Prod. 2019, 82, 3140–3149. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, A.; Tahaney, W.M.; Reddy Bollu, L.; Poage, G.; Hill, J.; Zhang, Y.; Mills, G.B.; Brown, P.H. The phosphatase PPM1A inhibits triple negative breast cancer growth by blocking cell cycle progression. NPJ. Breast Cancer 2019, 5, 22. [Google Scholar] [CrossRef]
- Yu, T.L.; Cai, D.L.; Zhu, G.F.; Ye, X.J.; Min, T.S.; Chen, H.Y.; Lu, D.R.; Chen, H.M. [Effects of CSN4 knockdown on proliferation and apoptosis of breast cancer MDA-MB-231 cells]. Yi Chuan 2019, 41, 318–326. [Google Scholar]
- Chen, J.; Ko, J.; Kim, J.T.; Cho, J.S.; Qiu, S.; Kim, G.D.; Auh, J.H.; Lee, H.J. beta-Thujaplicin inhibits basal-like mammary tumor growth by regulating glycogen synthase kinase-3beta/beta-catenin signaling. Food Funct. 2019, 10, 2691–2700. [Google Scholar] [CrossRef]
- Lee, S.O.; Lee, M.H.; Lee, K.R.; Lee, E.O.; Lee, H.J. Fomes fomentarius Ethanol Extract Exerts Inhibition of Cell Growth and Motility Induction of Apoptosis via Targeting AKT in Human Breast Cancer MDA-MB-231 Cells. Int. J. Mol. Sci. 2019, 20, 1147. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.C.; Hsieh, M.T.; Yang, J.S.; Lu, C.C.; Tsai, F.J.; Tsao, J.W.; Chiu, Y.J.; Kuo, S.C.; Lee, K.H. Effect of bis(hydroxymethyl) alkanoate curcuminoid derivative MTH-3 on cell cycle arrest, apoptotic and autophagic pathway in triple-negative breast adenocarcinoma MDA-MB-231 cells: An in vitro study. Int. J. Oncol. 2018, 52, 67–76. [Google Scholar] [CrossRef]
- Abd El-Hafeez, A.A.; Khalifa, H.O.; Mahdy, E.A.M.; Sharma, V.; Hosoi, T.; Ghosh, P.; Ozawa, K.; Montano, M.M.; Fujimura, T.; Ibrahim, A.R.N.; et al. Anticancer effect of nor-wogonin (5, 7, 8-trihydroxyflavone) on human triple-negative breast cancer cells via downregulation of TAK1, NF-kappaB, and STAT3. Pharmacol. Rep. 2019, 71, 289–298. [Google Scholar] [CrossRef]
- Liu, D.; You, P.; Luo, Y.; Yang, M.; Liu, Y. Galangin Induces Apoptosis in MCF-7 Human Breast Cancer Cells Through Mitochondrial Pathway and Phosphatidylinositol 3-Kinase/Akt Inhibition. Pharmacology 2018, 102, 58–66. [Google Scholar] [CrossRef]
- Bevara, G.B.; Naveen Kumar, A.D.; Koteshwaramma, K.L.; Badana, A.; Kumari, S.; Malla, R.R. C-glycosyl flavone from Urginea indica inhibits proliferation & angiogenesis & induces apoptosis via cyclin-dependent kinase 6 in human breast, hepatic & colon cancer cell lines. Indian J. Med. Res. 2018, 147, 158–168. [Google Scholar] [PubMed]
- Zhang, W.; Jiang, H.; Chen, Y.; Ren, F. Resveratrol chemosensitizes adriamycin-resistant breast cancer cells by modulating miR-122-5p. J. Cell Biochem. 2019, 120, 16283–16292. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Tan, S.; Duan, F.; Yuan, Q.; Li, Q.; Deng, G. Icariin induces apoptosis by suppressing autophagy in tamoxifen-resistant breast cancer cell line MCF-7/TAM. Breast Cancer 2019, 26, 766–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef]
- Hasanpourghadi, M.; Pandurangan, A.K.; Karthikeyan, C.; Trivedi, P.; Mustafa, M.R. Mechanisms of the anti-tumor activity of Methyl 2-(-5-fluoro-2-hydroxyphenyl)-1 H-benzo[d]imidazole-5-carboxylate against breast cancer in vitro and in vivo. Oncotarget 2017, 8, 28840–28853. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Lan, Z.; Xiong, X.; Ao, H.; Feng, Y.; Gu, H.; Yu, M.; Cui, Q. The Dual Role of MicroRNAs in Colorectal Cancer Progression. Int. J. Mol. Sci. 2018, 19, 2791. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Song, H.; Wu, T.; Li, D.; Hua, K.; Xu, H.; Zhao, B.; Wu, C.; Hu, J.; Ji, C.; et al. MicroRNA424 serves an antioncogenic role by targeting cyclindependent kinase 1 in breast cancer cells. Oncol. Rep. 2018, 40, 3416–3426. [Google Scholar]
- Zhao, J.; Li, D.; Fang, L. MiR-128-3p suppresses breast cancer cellular progression via targeting LIMK1. Biomed. Pharmacother 2019, 115, 108947. [Google Scholar] [CrossRef]
- Song, W.; Wu, S.; Wu, Q.; Zhou, L.; Yu, L.; Zhu, B.; Gong, X. The microRNA-141-3p/ CDK8 pathway regulates the chemosensitivity of breast cancer cells to trastuzumab. J. Cell Biochem. 2019, 120, 14095–14106. [Google Scholar] [CrossRef]
- Liu, L.H.; Tian, Q.Q.; Liu, J.; Zhou, Y.; Yong, H. Upregulation of hsa_circ_0136666 contributes to breast cancer progression by sponging miR-1299 and targeting CDK6. J. Cell Biochem. 2019, 120, 12684–12693. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, B.; Wang, X.; Zhang, F.; Yu, W. LINC00511 knockdown enhances paclitaxel cytotoxicity in breast cancer via regulating miR-29c/CDK6 axis. Life Sci. 2019, 228, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Cretella, D.; Fumarola, C.; Bonelli, M.; Alfieri, R.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Galetti, M.; Generali, D.; Petronini, P.G. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci. Rep. 2019, 9, 13014. [Google Scholar] [CrossRef] [PubMed]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M.; et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messer, J.A.; Ekinci, E.; Patel, T.A.; Teh, B.S. Enhanced dermatologic toxicity following concurrent treatment with palbociclib and radiation therapy: A case report. Rep. Pract. Oncol. Radiother 2019, 24, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Elango, R.; Vishnubalaji, R.; Manikandan, M.; Binhamdan, S.I.; Siyal, A.A.; Alshawakir, Y.A.; Alfayez, M.; Aldahmash, A.; Alajez, N.M. Concurrent targeting of BMI1 and CDK4/6 abrogates tumor growth in vitro and in vivo. Sci. Rep. 2019, 9, 13696. [Google Scholar] [CrossRef] [PubMed]
- DiPippo, A.J.; Patel, N.K.; Barnett, C.M. Cyclin-Dependent Kinase Inhibitors for the Treatment of Breast Cancer: Past, Present, and Future. Pharmacotherapy 2016, 36, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, T.J.; Meier, J.B.; Jansen, V.M. Current Landscape of Targeted Therapies for Hormone-Receptor Positive, HER2 Negative Metastatic Breast Cancer. Front. Oncol. 2018, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| CDKs | Partners | Established Functions | Biological Functions in BCs | Reference |
|---|---|---|---|---|
| CDK1 | Cyclin A/B | Associates with M phase of cell cycle | Associates with apoptosis of MYC-driven TNBC | [50,51,52] |
| CDK2 | Cyclin A/E | Associates with G1/S phase of cell cycle | Correlates with BC or TNBC phenotype | [52,53,54] |
| CDK3 | Cyclin C | Associates with G0/G1 and G1/S cell cycle transitions | Associates with BC cell migration, invasion, proliferation, and apoptosis | [27,55,56] |
| CDK4/6 | Cyclin D | Associates with the G1/S phase transition of the cell cycle | Contributes toward BC initiation and maintenance of tumorigenesis | [19,29] |
| CDK5 | p35 and p39 | Drives progression from G1/S and in RB phosphorylation; linked to chemotherapy resistance and immune response | Associates with ROS-mediated cell death in BC; essential for TGF-β1-induced epithelial–mesenchymal transition | [57,58,59,60,61] |
| CDK7 | Cyclin H | Associates with CAK and RNAPII transcription | Mediates transcriptional addiction to a vital cluster of genes in TNBC | [62,63] |
| CDK8 | Cyclin C | RNAPII transcription in complex; regulates the initiation of transcription | Responds to adjuvant therapy in BC; associated with tumor progression | [64,65,66,67] |
| CDK9 | Cyclin T | RNAPII transcription; promotes elongation of transcription | A prognostic biomarker in patients with BC following neoadjuvant chemotherapy | [45,68,69] |
| CDK10 | Cyclin M | Regulates ETS2 transcription, but not through RNAPII phosphorylation | Correlates with lymph node metastasis; resistance to endocrine therapy | [70,71,72] |
| CDK11 | Cyclin L | Regulates RNA transcription and splicing, autophagy, and apoptosis | Associates with growth and angiogenesis, proliferation, and apoptosis | [73,74,75,76,77] |
| CDK12 | Cyclin K | Controls alternative last exon splicing; regulates the expression of DNA damage, stress, and heat shock genes | Promotes BC cell invasion, an important therapeutic implication for TNBC; drives BC initiation and trastuzumab resistance | [47,78,79,80] |
| CDK13 | Cyclin K | Transcript synthesis toward the middle and 3′ end of the emerging RNA | Associated with DNA damage repair, genomic instability | [47,81,82] |
| CDK14 | Cyclin Y | Promotes Wnt/β-catenin signaling through phosphorylation of the LRP6 co-receptor | Associated with cell proliferation and invasion | [83,84,85,86] |
| CDK15 | Cyclin Y | Participates in hepatitis B virus-driven transformation | Associated with BC cell invasion and metastasis | [87,88] |
| CDK16 | Cyclin Y | Regulates mitosis, apoptosis, and growth; synaptic trafficking and remodeling | Associated with TRAIL | [89,90,91,92] |
| CDK17 | Cyclin Y | Promotes amyloid precursor protein-dependent Alzheimer; inhibits autophagy | Genetic expression profiles and chromosomal alterations | [93,94,95] |
| CDK18 | Cyclin Y | Promotes amyloid precursor protein-dependent Alzheimer; inhibits autophagy; promotes DNA replication stress and stability | Increases sensitivity to replication stress-inducing chemotherapeutic agents; induces DNA replication stress | [93,94,96,97,98] |
| CDK19 | Cyclin C | CDK8 paralog, with a similar role to CDK8, but seems to perform some distinct roles | The chemoresistance of BC; provides potential targets for the improving chemotherapy | [99,100] |
| CDK20 | Cyclin H | Activates ICK or β-catenin–TCF signaling to stimulate cell-cycle progression | The role of CDK20 needs to be further addressed in BC | [7,35] |
| Trial Name | Phase | Treatment Arms | Population | No. | PFS | Most Common AEs |
|---|---|---|---|---|---|---|
| PALOMA-1/ TRIO-18 (NCT00721409) | II | 1. Palbociclib + letrozole 2. Letrozole | Postmenopausal women, first-line treatment for ER+/HER2−ABC | 177 | 1. 20.2 months 2. 10.2 months | Neutropenia, leukopenia, fatigue anemia, nausea |
| PALOMA-2 (NCT01740427) | III | 1. Palbociclib + letrozole 2. Placebo + letrozole | ER+/HER2− ABC | 666 | 1. 19.3 months 2. 12.9 months | Neutropenia, leukopenia, nausea, fatigue, arthralgia, alopecia |
| PALOMA-3 (NCT01942135) | III | 1. Palbociclib + fulvestrant 2. Placebo + fulvestrant | HR+/HER2− ABC | 521 | 1. 9.2 months 2. 3.8 months | Neutropenia, leukopenia, thrombocytopenia, fatigue, nausea, headache, alopecia |
| PALLAS (NCT02513394) | III | 1. Palbociclib for 2 years + standard adjuvant ET for 5 years 2. Standard adjuvant ET for 5 years | ER+/HER2− early BC | 5795 | No detailed data | No detailed data |
| PENELOPE-B (NCT01864746) | III | 1. Palbociclib, 125 mg once daily, 28-day cycle for 13 cycles 2. Placebo 28-day cycle for 13 cycles | HR +/HER2− normal primary BC with high relapse risk after neoadjuvant chemotherapy | 1250 | No detailed data | No detailed data |
| MONALEESA-1 (NCT01919229) | II | 1. Letrozole + ribociclib. 2. Letrozole | Postmenopausal women with HR+/HER2− early BC | 14 | Mean decrease in Ki67-expressing cells, 1. 92%, 2. 69% | Nausea, decreased appetite, diarrhea, abdominal pain, fatigue, asthenia |
| MONALEESA-2 (NCT01958021) | III | 1. Ribociclib + letrozole 2. Placebo + letrozole | Postmenopausal women with HR+/HER2− MBC received no prior therapy for advanced disease | 668 | 1. 19.3 months 2. 14.7 months | Neutropenia, leukopenia, nausea, fatigue, diarrhea, alopecia |
| MONALEESA-3 (NCT02422615) | III | 1. Ribociclib + fulvestrant 2. Placebo + fulvestrant | Postmenopausal women with HR+/HER2− ABC received no or only one line prior endocrine treatment | 726 | 1. 20.5 months 2. 12.8 months | Neutropenia, leukopenia, nausea, fatigue, diarrhea, alopecia, vomiting, constipation, arthralgia, cough, headache, rash, anemia |
| MONALEESA-7 (NCT02278120) | III | 1. Ribociclib + NSAI/tamoxifen + goserelin 2. placebo + NSAI/tamoxifen + goserelin | Premenopausal or perimenopausal women with ER+/HER2−ABC | 672 | 1. 23.8 months 2. 13.0 months | Neutropenia, leukopenia, increased ALT, increased AST, anemia, hypertension |
| MONARCH-1 (NCT02102490) | II | Abemaciclib | heavily treated HR+/HER2− M/ABC patients (brain metastases were excluded) | 132 | 6.0 months (95% confidence interval (CI) 4.2 to 7.5) | Leucopenia, neutropenia, diarrhea, fatigue, nausea, hypokalemia, increased ALT, decreased appetite, hyponatremia, abdominal pain, thrombocytopenia |
| MONARCH-2 (NCT02107703) | III | 1. Abemaciclib + fulvestrant 2. Placebo + fulvestrant | HR+/HER2− locally advanced or metastatic BC. | 669 | 1. 16.4 months 2. 9.3 months | Neutropenia, diarrhea, nausea, fatigue, abdominal pain |
| MONARCH-3 (NCT02246621) | III | 1. Abemaciclib + anastrozole/ letrozole 2. Placebo + anastrozole/ letrozole | Postmenopausal women HR+/HER2− locoregionally, recurrent, or MBC | 493 | 1. 28.2 months 2. 14.8 months | Neutropenia, diarrhea, nausea, fatigue, infections |
| Trial Name | Phase | Status | Design | Treatment Arms | Population | Pts Enrolled | Objectives |
|---|---|---|---|---|---|---|---|
| NCT02947685 | III | R | Randomized, parallel assignment, open label | 1. Palbociclib + anti-HER2 therapy (trastuzumab/pertuzumab) + ET (letrozole, anastrozole, exemestane, fulvestrant) 2. Anti-HER2 therapy (trastuzumab/pertuzumab) + ET (letrozole, anastrozole, exemestane, fulvestrant) | HER2+/ER+ BC | 496 (estimated) | PFS, OS, ORR, DOR, CBR, safety, 3 and 5 year survival probabilities |
| NCT02774681 | II | Terminated | Single group assignment, open label | 1. palbociclib PO 2. palbociclib PO + trastuzumab IV | HER2+/PR− MBC with brain metastasis | 12 (estimated) | AEs, CNS, PFS, OS, CNS, ORR, safety, tolerability |
| NCT02530424 | II | Active, N/R | Single group assignment, open label | (Trastuzumab + Pertuzumab + Palbociclib ± Fulvestrant) + Surgery | ER+/HER2+ BC suitable for neoadjuvant therapy | 102 (actual) | PCR, COR, safety, tolerability |
| NCT02657343 | Ib/II | Active, N/R | Non- randomized, parallel assignment, open label | 1. Ribociclib + T-DM1 2. Ribociclib + Trastuzumab 3. Ribociclib + Trastuzumab + Fulvestrant | HER+ A/MBC | 26 (actual) | Mtd, RP2D,CBR, ORR, PFS, OS. |
| NCT03913234 | I/II | Not yet R | Single group assignment, open label | Ribociclib + Trastuzumab + Letrozole | Postmenopausal HER2+ MBC | 95 (estimated) | PFS,OS, RT, QOL |
| NCT03054363 | Ib/II | R | Non- randomized, single group assignment, open label | Tucatinib + Palbociclib + Letrozole | HR+/HER2+ A/MBC | 25 (estimated) | AEs, PFS |
| NCT03993964 | II | Not yet R | Single group assignment, open label | Pyrotinib + SHR6390 | HER2+ ABC | 20 (estimated) | ORR, PFS, OS |
| NCT03090165 | I/II | Active,N/R | Single group assignment, open label | 1. bicalutamide + ribociclib 400mg PO daily on days 1-21 of a 28-day cycle. 2. bicalutamide + ribociclib 400mg PO daily on days 1-28 of a 28-day cycle. 3. bicalutamide + ribociclib 600mg PO daily on days 1-21 of a 28-day cycle. | AR+ TNBC | 11 (actual) | ORR, DOR, safety, tolerability, PFS, OS, CBR, |
| NCT03805399 | Ib/II | R | Non- randomized, open label, umbrella study, parallel assignment | 1. Pyrotinib + Capecitabine 2. AR inhibitor + CDK4/6 inhibitor 3. anti PD-1 + nab-paclitaxel 4. PARP inhibitor 5. BLIS + anti-VEGFR 6. MES + anti-VEGFR 7. mTOR inhibitor + nab-paclitaxel | TNBC | 140 (estimated) | ORR, DOR, PFS, OS |
| NCT03519178 | II | R | Non- randomized, single group assignment, open label | 1. PF-06873600 2. PF-06873600 + Endocrine Therapy 1 3. PF-06873600 + Endocrine Therapy 2 | HR+/HER2− MBC, TNBC | 220 (estimated) | DL, safety, tolerability, ORR, Cmax, Tmax, PK |
| NCT02907918 | II | R | Single group assignment, open label | (Palbociclib + letrozole + trastuzumab +/- goserelin) + surgery | ER+/HER2+ Stage II-III BC | 48 (estimated) | PCR, safety, tolerability |
| NCT02605486 | I/II | R | Single group assignment, non- randomized, open label | Palbociclib + Bicalutamide | AR+/ER− MBC | 51 (estimated) | RP2D, PFS, ORR, CBR, safety, tolerability |
| NCT02675231 | II | Active, N/R | Randomized, parallel assignment, open label | 1. Abemaciclib + Trastuzumab + Fulvestrant 2. Abemaciclib + Trastuzumab 3. Trastuzumab + Standard of Care Chemotherapy | HR+/HER2+ A/MBC | 225 (estimated) | PFS, OS, CR, PR, DOR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. https://doi.org/10.3390/ijms21061960
Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, Yu M, Lin J, Cui Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. International Journal of Molecular Sciences. 2020; 21(6):1960. https://doi.org/10.3390/ijms21061960
Chicago/Turabian StyleDing, Lei, Jiaqi Cao, Wen Lin, Hongjian Chen, Xianhui Xiong, Hongshun Ao, Min Yu, Jie Lin, and Qinghua Cui. 2020. "The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer" International Journal of Molecular Sciences 21, no. 6: 1960. https://doi.org/10.3390/ijms21061960
APA StyleDing, L., Cao, J., Lin, W., Chen, H., Xiong, X., Ao, H., Yu, M., Lin, J., & Cui, Q. (2020). The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. International Journal of Molecular Sciences, 21(6), 1960. https://doi.org/10.3390/ijms21061960