3-Iodothyronamine and Derivatives: New Allies Against Metabolic Syndrome?





Abstract
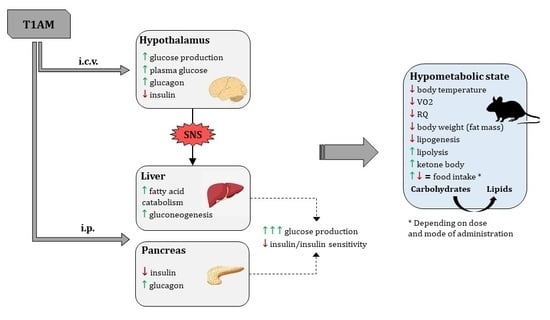
:
1. Introduction
2. Thyronamine: A Novel Pathway of Metabolic Regulation
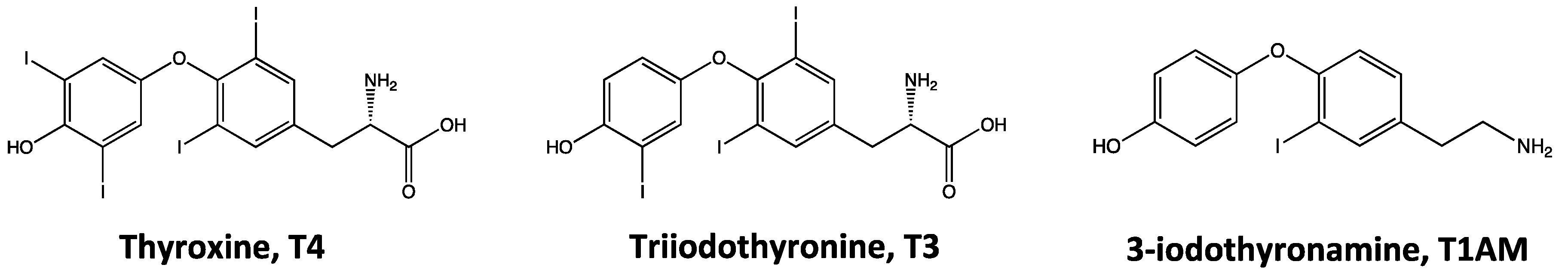
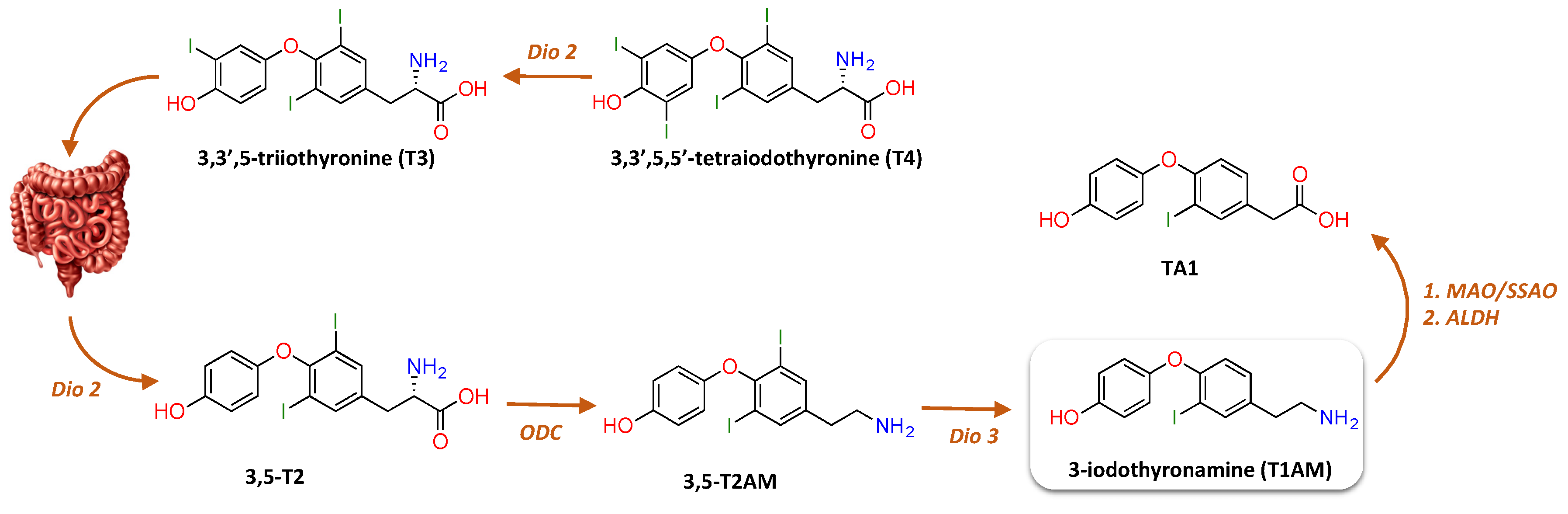
2.1. Endogenous Biosynthesis and Biodistribution of 3-Iodothyronamine (T1AM)
2.2. Thyronamine Regulation of Metabolism
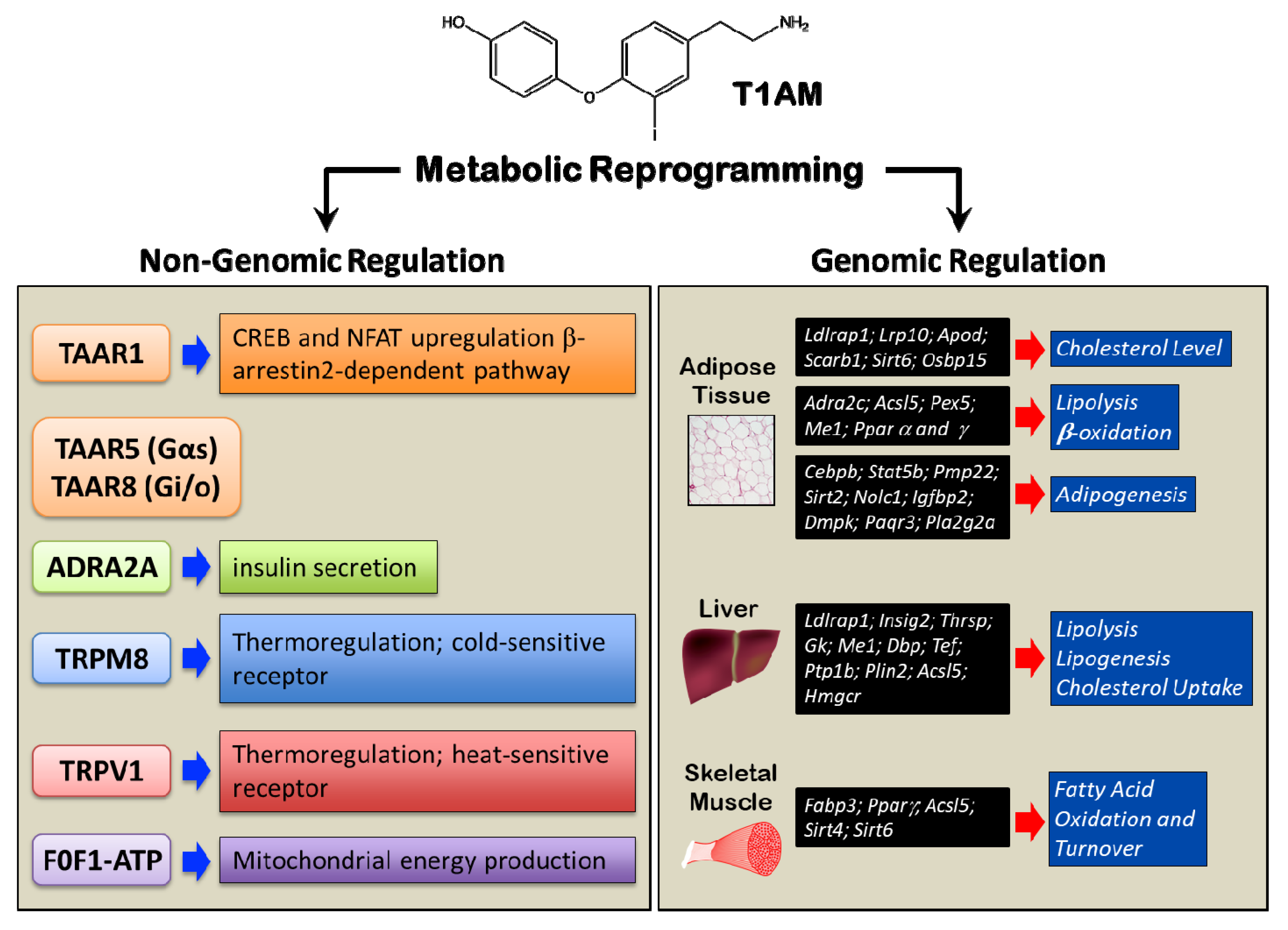
2.3. Molecular Targets of Non-Genomic Regulation of Metabolism
2.4. Genomic Regulation of Metabolism
3. Pathophysiological Implications
4. Therapeutic Implications and Development Direction
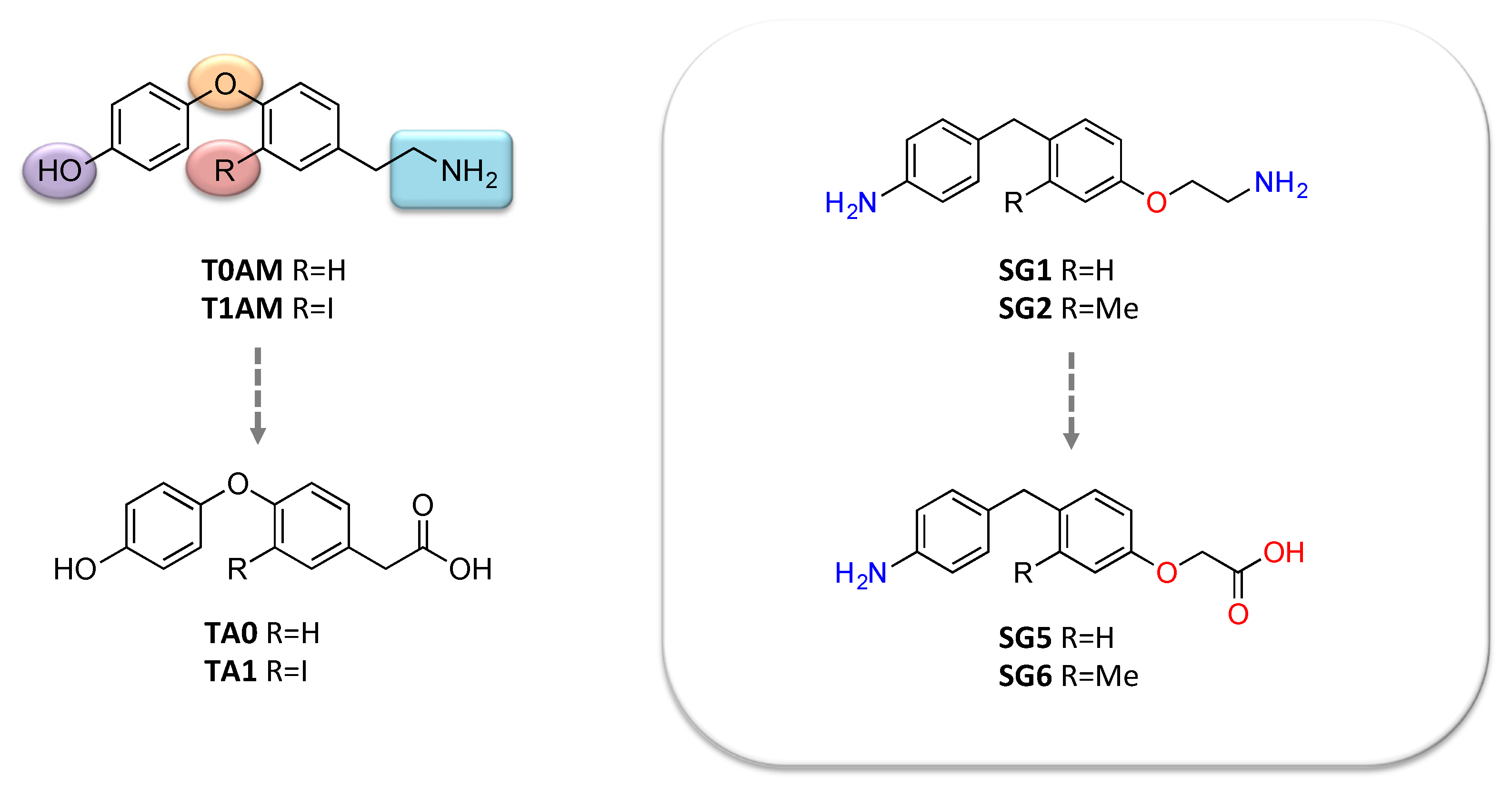
5. Novel 3-Iodothyronamine Derivatives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hennekens, C.H.; Andreotti, F. Leading avoidable cause of premature deaths worldwide: Case for obesity. Am. J. Med. 2013, 126, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Galassi, A.; Reynolds, K.; He, J. Metabolic syndrome and risk of cardiovascular disease: A meta-analysis. Am. J. Med. 2006, 119, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.; Okoloise, M.; Williams, K.; Stern, M.P.; Haffner, S.M.; San Antonio Heart, S. The metabolic syndrome as predictor of type 2 diabetes: The San Antonio heart study. Diabetes Care 2003, 26, 3153–3159. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef] [Green Version]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States, 2003–2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Group, D.S. Does the constellation of risk factors with and without abdominal adiposity associate with different cardiovascular mortality risk? Int. J. Obes. (Lond.) 2008, 32, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Mabry, R.M.; Reeves, M.M.; Eakin, E.G.; Owen, N. Gender differences in prevalence of the metabolic syndrome in Gulf Cooperation Council Countries: A systematic review. Diabet Med. 2010, 27, 593–597. [Google Scholar] [CrossRef]
- Gennuso, K.P.; Gangnon, R.E.; Thraen-Borowski, K.M.; Colbert, L.H. Dose-response relationships between sedentary behaviour and the metabolic syndrome and its components. Diabetologia 2015, 58, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, R.; Sullivan, L.; Jacques, P.F.; Wang, T.J.; Fox, C.S.; Meigs, J.B.; D’Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 2007, 116, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Green, A.K.; Jacques, P.F.; Rogers, G.; Fox, C.S.; Meigs, J.B.; McKeown, N.M. Sugar-sweetened beverages and prevalence of the metabolically abnormal phenotype in the Framingham Heart Study. Obesity (Silver Spring) 2014, 22, E157–E163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsey, P.L.; Steffen, L.M.; Stevens, J. Dietary intake and the development of the metabolic syndrome: The Atherosclerosis Risk in Communities study. Circulation 2008, 117, 754–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef]
- Tooke, J.E.; Hannemann, M.M. Adverse endothelial function and the insulin resistance syndrome. J. Intern. Med. 2000, 247, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Juhan-Vague, I.; Alessi, M.C.; Mavri, A.; Morange, P.E. Plasminogen activator inhibitor-1, inflammation, obesity, insulin resistance and vascular risk. J. Thromb. Haemost. 2003, 1, 1575–1579. [Google Scholar] [CrossRef]
- Wallace, A.M.; McMahon, A.D.; Packard, C.J.; Kelly, A.; Shepherd, J.; Gaw, A.; Sattar, N. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation 2001, 104, 3052–3056. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Al-Adsani, H.; Hoffer, L.J.; Silva, J.E. Resting energy expenditure is sensitive to small dose changes in patients on chronic thyroid hormone replacement. J. Clin. Endocrinol. Metab. 1997, 82, 1118–1125. [Google Scholar] [CrossRef]
- Motomura, K.; Brent, G.A. Mechanisms of thyroid hormone action. Implications for the clinical manifestation of thyrotoxicosis. Endocrinol. Metab. Clin. N. Am. 1998, 27, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Delitala, A.P.; Fanciulli, G.; Pes, G.M.; Maioli, M.; Delitala, G. Thyroid Hormones, Metabolic Syndrome and Its Components. Endocr. Metab. Immune Disord. Drug Targets 2017, 17, 56–62. [Google Scholar] [CrossRef] [PubMed]
- De Meis, L. Role of the sarcoplasmic reticulum Ca2+-ATPase on heat production and thermogenesis. Biosci. Rep. 2001, 21, 113–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.E. Thermogenic mechanisms and their hormonal regulation. Physiol. Rev. 2006, 86, 435–464. [Google Scholar] [CrossRef]
- Harper, M.E.; Brand, M.D. The quantitative contributions of mitochondrial proton leak and ATP turnover reactions to the changed respiration rates of hepatocytes from rats of different thyroid status. J. Biol. Chem. 1993, 268, 14850–14860. [Google Scholar]
- Sacks, H.; Symonds, M.E. Anatomical locations of human brown adipose tissue: Functional relevance and implications in obesity and type 2 diabetes. Diabetes 2013, 62, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [Green Version]
- Fliers, E.; Klieverik, L.P.; Kalsbeek, A. Novel neural pathways for metabolic effects of thyroid hormone. Trends Endocrinol. Metab. 2010, 21, 230–236. [Google Scholar] [CrossRef]
- Kouidhi, S.; Clerget-Froidevaux, M.S. Integrating Thyroid Hormone Signaling in Hypothalamic Control of Metabolism: Crosstalk Between Nuclear Receptors. Int. J. Mol. Sci. 2018, 19, 2017. [Google Scholar] [CrossRef] [Green Version]
- Krotkiewski, M. Thyroid hormones in the pathogenesis and treatment of obesity. Eur. J. Pharmacol. 2002, 440, 85–98. [Google Scholar] [CrossRef]
- Hoefig, C.S.; Wuensch, T.; Rijntjes, E.; Lehmphul, I.; Daniel, H.; Schweizer, U.; Mittag, J.; Kohrle, J. Biosynthesis of 3-Iodothyronamine From T4 in Murine Intestinal Tissue. Endocrinology 2015, 156, 4356–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piehl, S.; Heberer, T.; Balizs, G.; Scanlan, T.S.; Smits, R.; Koksch, B.; Kohrle, J. Thyronamines are isozyme-specific substrates of deiodinases. Endocrinology 2008, 149, 3037–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanlan, T.S.; Suchland, K.L.; Hart, M.E.; Chiellini, G.; Huang, Y.; Kruzich, P.J.; Frascarelli, S.; Crossley, D.A.; Bunzow, J.R.; Ronca-Testoni, S.; et al. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat. Med. 2004, 10, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, G.; Frascarelli, S.; Ghelardoni, S.; Carnicelli, V.; Tobias, S.C.; DeBarber, A.; Brogioni, S.; Ronca-Testoni, S.; Cerbai, E.; Grandy, D.K.; et al. Cardiac effects of 3-iodothyronamine: A new aminergic system modulating cardiac function. FASEB J. 2007, 21, 1597–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, A.; Chiellini, G.; Frascarelli, S.; Marchini, M.; Ghelardoni, S.; Raffaelli, A.; Tonacchera, M.; Vitti, P.; Scanlan, T.S.; Zucchi, R. Tissue distribution and cardiac metabolism of 3-iodothyronamine. Endocrinology 2010, 151, 5063–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, S.S.; Sniegoski, L.T.; Welch, M.J. Candidate reference method for total thyroxine in human serum: Use of isotope-dilution liquid chromatography-mass spectrometry with electrospray ionization. Clin. Chem. 2002, 48, 637–642. [Google Scholar] [CrossRef]
- Tai, S.S.; Bunk, D.M.; White, E.T.; Welch, M.J. Development and evaluation of a reference measurement procedure for the determination of total 3,3′,5-triiodothyronine in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal. Chem. 2004, 76, 5092–5096. [Google Scholar] [CrossRef]
- Ackermans, M.T.; Klieverik, L.P.; Ringeling, P.; Endert, E.; Kalsbeek, A.; Fliers, E. An online solid-phase extraction-liquid chromatography-tandem mass spectrometry method to study the presence of thyronamines in plasma and tissue and their putative conversion from 13C6-thyroxine. J. Endocrinol. 2010, 206, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Hoefig, C.S.; Kohrle, J.; Brabant, G.; Dixit, K.; Yap, B.; Strasburger, C.J.; Wu, Z. Evidence for extrathyroidal formation of 3-iodothyronamine in humans as provided by a novel monoclonal antibody-based chemiluminescent serum immunoassay. J. Clin. Endocrinol. Metab. 2011, 96, 1864–1872. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.E.; De Siena, G.; Saba, A.; Marchini, M.; Landucci, E.; Gerace, E.; Zazzeri, M.; Musilli, C.; Pellegrini-Giampietro, D.; Matucci, R.; et al. Pharmacological effects of 3-iodothyronamine (T1AM) in mice include facilitation of memory acquisition and retention and reduction of pain threshold. Br. J. Pharmacol. 2013, 168, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Assadi-Porter, F.M.; Reiland, H.; Sabatini, M.; Lorenzini, L.; Carnicelli, V.; Rogowski, M.; Selen Alpergin, E.S.; Tonelli, M.; Ghelardoni, S.; Saba, A.; et al. Metabolic Reprogramming by 3-Iodothyronamine (T1AM): A New Perspective to Reverse Obesity through Co-Regulation of Sirtuin 4 and 6 Expression. Int. J. Mol. Sci. 2018, 19, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, G.; Placzek, E.; Scanlan, T.S. ApoB-100-containing lipoproteins are major carriers of 3-iodothyronamine in circulation. J. Biol. Chem. 2012, 287, 1790–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braulke, L.J.; Klingenspor, M.; DeBarber, A.; Tobias, S.C.; Grandy, D.K.; Scanlan, T.S.; Heldmaier, G. 3-Iodothyronamine: A novel hormone controlling the balance between glucose and lipid utilisation. J. Comp. Physiol. B 2008, 178, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Selen Alpergin, E.S.; Bolandnazar, Z.; Sabatini, M.; Rogowski, M.; Chiellini, G.; Zucchi, R.; Assadi-Porter, F.M. Metabolic profiling reveals reprogramming of lipid metabolic pathways in treatment of polycystic ovary syndrome with 3-iodothyronamine. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Hettinger, B.D.; Schuff, K.; Marks, D.; Scanlan, T.S. 3-Iodothyronamine (T1AM) causes weight loss in mice via reduction in food consumption. In Proceedings of the14th International Thyroid Congress, Paris, France, 11–16 September 2010. OC-141. [Google Scholar]
- Haviland, J.A.; Reiland, H.; Butz, D.E.; Tonelli, M.; Porter, W.P.; Zucchi, R.; Scanlan, T.S.; Chiellini, G.; Assadi-Porter, F.M. NMR-based metabolomics and breath studies show lipid and protein catabolism during low dose chronic T(1)AM treatment. Obesity (Silver Spring) 2013, 21, 2538–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gachkar, S.; Oelkrug, R.; Martinez-Sanchez, N.; Rial-Pensado, E.; Warner, A.; Hoefig, C.S.; Lopez, M.; Mittag, J. 3-Iodothyronamine Induces Tail Vasodilation Through Central Action in Male Mice. Endocrinology 2017, 158, 1977–1984. [Google Scholar] [CrossRef]
- Dhillo, W.S.; Bewick, G.A.; White, N.E.; Gardiner, J.V.; Thompson, E.L.; Bataveljic, A.; Murphy, K.G.; Roy, D.; Patel, N.A.; Scutt, J.N.; et al. The thyroid hormone derivative 3-iodothyronamine increases food intake in rodents. Diabetes Obes. Metab. 2009, 11, 251–260. [Google Scholar] [CrossRef]
- Manni, M.E.; De Siena, G.; Saba, A.; Marchini, M.; Dicembrini, I.; Bigagli, E.; Cinci, L.; Lodovici, M.; Chiellini, G.; Zucchi, R.; et al. 3-Iodothyronamine: A modulator of the hypothalamus-pancreas-thyroid axes in mice. Br. J. Pharmacol. 2012, 166, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Eskandarzade, N.; Kazemipour, N.; Jafarizade, A.; Nazifi, S. The Possible Role of 3-lodothyronamine in Browning of Inguinal White Adipose Tissue in Mice. Turk. J. Endocrinol. Metab. 2017, 21, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Klieverik, L.P.; Foppen, E.; Ackermans, M.T.; Serlie, M.J.; Sauerwein, H.P.; Scanlan, T.S.; Grandy, D.K.; Fliers, E.; Kalsbeek, A. Central effects of thyronamines on glucose metabolism in rats. J. Endocrinol. 2009, 201, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Regard, J.B.; Kataoka, H.; Cano, D.A.; Camerer, E.; Yin, L.; Zheng, Y.W.; Scanlan, T.S.; Hebrok, M.; Coughlin, S.R. Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J. Clin. Investig. 2007, 117, 4034–4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghelardoni, S.; Chiellini, G.; Frascarelli, S.; Saba, A.; Zucchi, R. Uptake and metabolic effects of 3-iodothyronamine in hepatocytes. J. Endocrinol. 2014, 221, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowsky, B. Trace amines: Identification of a family of mammalian G protein–coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunzow, J.R. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 2001, 60. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, L.; Ebeling, M.; Kratochwil, N.A.; Bunzow, J.R.; Grandy, D.K.; Hoener, M.C. Trace amine-associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors. Genomics 2005, 85, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Rutigliano, G.; Accorroni, A.; Zucchi, R. The Case for TAAR1 as a Modulator of Central Nervous System Function. Front. Pharmacol. 2017, 8, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchi, R.; Chiellini, G.; Scanlan, T.S.; Grandy, D.K. Trace amine-associated receptors and their ligands. Br. J. Pharmacol. 2006, 149, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Coster, M.; Biebermann, H.; Schoneberg, T.; Staubert, C. Evolutionary Conservation of 3-Iodothyronamine as an Agonist at the Trace Amine-Associated Receptor 1. Eur. Thyroid J. 2015, 4, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Asif-Malik, A.; Canales, J.J. Trace Amines and the Trace Amine-Associated Receptor 1: Pharmacology, Neurochemistry, and Clinical Implications. Front. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Laurino, A.; Landucci, E.; Resta, F.; De Siena, G.; Pellegrini-Giampietro, D.E.; Masi, A.; Mannaioni, G.; Raimondi, L. Anticonvulsant and Neuroprotective Effects of the Thyroid Hormone Metabolite 3-Iodothyroacetic Acid. Thyroid 2018, 28, 1387–1397. [Google Scholar] [CrossRef]
- Panas, H.N.; Lynch, L.J.; Vallender, E.J.; Xie, Z.; Chen, G.L.; Lynn, S.K.; Scanlan, T.S.; Miller, G.M. Normal thermoregulatory responses to 3-iodothyronamine, trace amines and amphetamine-like psychostimulants in trace amine associated receptor 1 knockout mice. J. Neurosci. Res. 2010, 88, 1962–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinau, G.; Pratzka, J.; Nurnberg, D.; Gruters, A.; Fuhrer-Sakel, D.; Krude, H.; Kohrle, J.; Schoneberg, T.; Biebermann, H. Differential modulation of Beta-adrenergic receptor signaling by trace amine-associated receptor 1 agonists. PLoS ONE 2011, 6, e27073. [Google Scholar] [CrossRef] [PubMed]
- Dinter, J.; Muhlhaus, J.; Wienchol, C.L.; Yi, C.X.; Nurnberg, D.; Morin, S.; Gruters, A.; Kohrle, J.; Schoneberg, T.; Tschop, M.; et al. Inverse agonistic action of 3-iodothyronamine at the human trace amine-associated receptor 5. PLoS ONE 2015, 10, e0117774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiellini, G.; Erba, P.; Carnicelli, V.; Manfredi, C.; Frascarelli, S.; Ghelardoni, S.; Mariani, G.; Zucchi, R. Distribution of exogenous [125I]-3-iodothyronamine in mouse in vivo: Relationship with trace amine-associated receptors. J. Endocrinol. 2012, 213, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Ito, M.; Nambu, H.; Fujikawa, T.; Tanaka, K.; Iwaasa, H.; Tokita, S. Anatomical and histological profiling of orphan G-protein-coupled receptor expression in gastrointestinal tract of C57BL/6J mice. Cell Tissue Res. 2009, 338, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Kunnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol. Metab. 2016, 5, 47–56. [Google Scholar] [CrossRef]
- James, T.D.; Moffett, S.X.; Scanlan, T.S.; Martin, J.V. Effects of acute microinjections of the thyroid hormone derivative 3-iodothyronamine to the preoptic region of adult male rats on sleep, thermoregulation and motor activity. Horm. Behav. 2013, 64, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Lucius, A.; Khajavi, N.; Reinach, P.S.; Kohrle, J.; Dhandapani, P.; Huimann, P.; Ljubojevic, N.; Grotzinger, C.; Mergler, S. 3-Iodothyronamine increases transient receptor potential melastatin channel 8 (TRPM8) activity in immortalized human corneal epithelial cells. Cell. Signal. 2016, 28, 136–147. [Google Scholar] [CrossRef]
- Khajavi, N.; Reinach, P.S.; Slavi, N.; Skrzypski, M.; Lucius, A.; Strauss, O.; Kohrle, J.; Mergler, S. Thyronamine induces TRPM8 channel activation in human conjunctival epithelial cells. Cell. Signal. 2015, 27, 315–325. [Google Scholar] [CrossRef]
- Khajavi, N.; Mergler, S.; Biebermann, H. 3-Iodothyronamine, a Novel Endogenous Modulator of Transient Receptor Potential Melastatin 8? Front. Endocrinol. (Lausanne) 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Fang, Y.; Ronnekleiv, O.K.; Kelly, M.J. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J. Neurosci. 2010, 30, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.M.; McAlexander, M.A.; Biro, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef]
- Sukumar, P.; Sedo, A.; Li, J.; Wilson, L.A.; O’Regan, D.; Lippiat, J.D.; Porter, K.E.; Kearney, M.T.; Ainscough, J.F.; Beech, D.J. Constitutively active TRPC channels of adipocytes confer a mechanism for sensing dietary fatty acids and regulating adiponectin. Circ. Res. 2012, 111, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Kunert-Keil, C.; Bisping, F.; Kruger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunig, J.; Mergler, S.; Jyrch, S.; Hoefig, C.S.; Rosowski, M.; Mittag, J.; Biebermann, H.; Khajavi, N. 3-Iodothyronamine Activates a Set of Membrane Proteins in Murine Hypothalamic Cell Lines. Front. Endocrinol. (Lausanne) 2018, 9, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianculescu, A.G.; Giacomini, K.M.; Scanlan, T.S. Identification and characterization of 3-iodothyronamine intracellular transport. Endocrinology 2009, 150, 1991–1999. [Google Scholar] [CrossRef] [Green Version]
- Cumero, S.; Fogolari, F.; Domenis, R.; Zucchi, R.; Mavelli, I.; Contessi, S. Mitochondrial F(0) F(1) -ATP synthase is a molecular target of 3-iodothyronamine, an endogenous metabolite of thyroid hormone. Br. J. Pharmacol. 2012, 166, 2331–2347. [Google Scholar] [CrossRef] [Green Version]
- Venditti, P.; Napolitano, G.; Di Stefano, L.; Chiellini, G.; Zucchi, R.; Scanlan, T.S.; Di Meo, S. Effects of the thyroid hormone derivatives 3-iodothyronamine and thyronamine on rat liver oxidative capacity. Mol. Cell. Endocrinol. 2011, 341, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Lehmphul, I.; Hoefig, C.S.; Kohrle, J. 3-Iodothyronamine reduces insulin secretion in vitro via a mitochondrial mechanism. Mol. Cell. Endocrinol. 2018, 460, 219–228. [Google Scholar] [CrossRef]
- Mariotti, V.; Melissari, E.; Iofrida, C.; Righi, M.; Di Russo, M.; Donzelli, R.; Saba, A.; Frascarelli, S.; Chiellini, G.; Zucchi, R.; et al. Modulation of gene expression by 3-iodothyronamine: Genetic evidence for a lipolytic pattern. PLoS ONE 2014, 9, e106923. [Google Scholar] [CrossRef] [Green Version]
- Guarente, L. Sirtuins as potential targets for metabolic syndrome. Nature 2006, 444, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.; Yunis, E.J.; Good, R.A. Suppression of adenocarcinoma by the immunological consequences of calorie restriction. Nature 1976, 263, 504–507. [Google Scholar] [CrossRef]
- Zhu, H.; Guo, Q.; Mattson, M.P. Dietary restriction protects hippocampal neurons against the death-promoting action of a presenilin-1 mutation. Brain Res. 1999, 842, 224–229. [Google Scholar] [CrossRef]
- Ingram, D.K.; Weindruch, R.; Spangler, E.L.; Freeman, J.R.; Walford, R.L. Dietary restriction benefits learning and motor performance of aged mice. J. Gerontol. 1987, 42, 78–81. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Bellusci, L.; Laurino, A.; Sabatini, M.; Sestito, S.; Lenzi, P.; Raimondi, L.; Rapposelli, S.; Biagioni, F.; Fornai, F.; Salvetti, A.; et al. New Insights into the Potential Roles of 3-Iodothyronamine (T1AM) and Newly Developed Thyronamine-Like TAAR1 Agonists in Neuroprotection. Front. Pharmacol. 2017, 8, 905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accorroni, A.; Rutigliano, G.; Sabatini, M.; Frascarelli, S.; Borso, M.; Novelli, E.; Bandini, L.; Ghelardoni, S.; Saba, A.; Zucchi, R.; et al. Exogenous 3-Iodothyronamine Rescues the Entorhinal Cortex from beta-Amyloid Toxicity. Thyroid 2019. [Google Scholar] [CrossRef]
- Frascarelli, S.; Ghelardoni, S.; Chiellini, G.; Galli, E.; Ronca, F.; Scanlan, T.S.; Zucchi, R. Cardioprotective effect of 3-iodothyronamine in perfused rat heart subjected to ischemia and reperfusion. Cardiovasc. Drugs Ther. 2011, 25, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, F.; Rutigliano, G.; Borsò, M.; Chiellini, G.; Saba, A.; Origlia, N.; Zucchi, R. Release of endogenous 3-iodothyroacetic acid in a mouse model of short-term brain ischemia. In Proceedings of the 42nd Annual Meeting of the European Thyroid Association, Budapest, Hungary, 7–10 September 2019. [Google Scholar]
- Hackenmueller, S.A.; Marchini, M.; Saba, A.; Zucchi, R.; Scanlan, T.S. Biosynthesis of 3-iodothyronamine (T1AM) is dependent on the sodium-iodide symporter and thyroperoxidase but does not involve extrathyroidal metabolism of T4. Endocrinology 2012, 153, 5659–5667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, E.; Marchini, M.; Saba, A.; Berti, S.; Tonacchera, M.; Vitti, P.; Scanlan, T.S.; Iervasi, G.; Zucchi, R. Detection of 3-iodothyronamine in human patients: A preliminary study. J. Clin. Endocrinol. Metab. 2012, 97, E69–E74. [Google Scholar] [CrossRef] [PubMed]
- Langouche, L.; Lehmphul, I.; Perre, S.V.; Kohrle, J.; Van den Berghe, G. Circulating 3-T1AM and 3,5-T2 in Critically Ill Patients: A Cross-Sectional Observational Study. Thyroid 2016, 26, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- La Cour, J.L.; Christensen, H.M.; Kohrle, J.; Lehmphul, I.; Kistorp, C.; Nygaard, B.; Faber, J. Association Between 3-Iodothyronamine (T1am) Concentrations and Left Ventricular Function in Chronic Heart Failure. J. Clin. Endocrinol. Metab. 2019, 104, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Muhlhaus, J.; Dinter, J.; Jyrch, S.; Teumer, A.; Jacobi, S.F.; Homuth, G.; Kuhnen, P.; Wiegand, S.; Gruters, A.; Volzke, H.; et al. Investigation of Naturally Occurring Single-Nucleotide Variants in Human TAAR1. Front. Pharmacol. 2017, 8, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucchi, R.; Rutigliano, G.; Saponaro, F. Novel thyroid hormones. Endocrine 2019, 66, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, G.; Bellusci, L.; Sabatini, M.; Zucchi, R. Thyronamines and Analogues—The Route from Rediscovery to Translational Research on Thyronergic Amines. Mol. Cell. Endocrinol. 2017, 458, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Virili, C.; Centanni, M. With a little help from my friends—The role of microbiota in thyroid hormone metabolism and enterohepatic recycling. Mol. Cell. Endocrinol. 2017, 458, 39–43. [Google Scholar] [CrossRef]
- Cichero, E. Opportunities and Challenges in the Design of Selective TAAR1 Agonists: An Editorial; Taylor & Francis: Abingdon, UK, 2018. [Google Scholar]
- Hart, M.E.; Suchland, K.L.; Miyakawa, M.; Bunzow, J.R.; Grandy, D.K.; Scanlan, T.S. Trace amine-associated receptor agonists: Synthesis and evaluation of thyronamines and related analogues. J. Med. Chem. 2006, 49, 1101–1112. [Google Scholar] [CrossRef]
- Tan, E.S.; Miyakawa, M.; Bunzow, J.R.; Grandy, D.K.; Scanlan, T.S. Exploring the Structure—Activity Relationship of the Ethylamine Portion of 3-Iodothyronamine for Rat and Mouse Trace Amine-Associated Receptor 1. J. Med. Chem. 2007, 50, 2787–2798. [Google Scholar] [CrossRef]
- Chiellini, G.; Nesi, G.; Digiacomo, M.; Malvasi, R.; Espinoza, S.; Sabatini, M.; Frascarelli, S.; Laurino, A.; Cichero, E.; Macchia, M. Design, Synthesis, and Evaluation of Thyronamine Analogues as Novel Potent Mouse Trace Amine Associated Receptor 1 (m TAAR1) Agonists. J. Med. Chem. 2015, 58, 5096–5107. [Google Scholar] [CrossRef]
- Chiellini, G.; Nesi, G.; Sestito, S.; Chiarugi, S.; Runfola, M.; Espinoza, S.; Sabatini, M.; Bellusci, L.; Laurino, A.; Cichero, E. Hit-to-lead optimization of mouse Trace Amine Associated Receptor 1 (mTAAR1) agonists with a diphenylmethane-scaffold: Design, Synthesis, and biological study. J. Med. Chem. 2016, 59, 9825–9836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogowski, M.; Bellusci, L.; Sabatini, M.; Rapposelli, S.; Rahman, S.M.; Chiellini, G.; Assadi-Porter, F.M. Lipolytic Effects of 3-Iodothyronamine (T1AM) and a Novel Thyronamine-Like Analog SG-2 through the AMPK Pathway. Int. J. Mol. Sci. 2019, 20, 4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogowski, M.; Gollahon, L.; Chellini, G.; Assadi-Porter, F.M. Uptake of 3-iodothyronamine hormone analogs inhibits the growth and viability of cancer cells. FEBS Open Bio 2017, 7, 587–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musilli, C.; De Siena, G.; Manni, M.E.; Logli, A.; Landucci, E.; Zucchi, R.; Saba, A.; Donzelli, R.; Passani, M.B.; Provensi, G. Histamine mediates behavioural and metabolic effects of 3-iodothyroacetic acid, an endogenous end product of thyroid hormone metabolism. Br. J. Pharmacol. 2014, 171, 3476–3484. [Google Scholar] [CrossRef] [Green Version]
- Bellusci, L.; Runfola, M.; Carnicelli, V.; Sestito, S.; Fulceri, F.; Santucci, F.; Lenzi, P.; Fornai, F.; Rapposelli, S.; Origlia, N.; et al. Endogenous 3-Iodothyronamine (T1AM) and Synthetic Thyronamine-like Analog SG-2 Act as Novel Pleiotropic Neuroprotective Agents Through the Modulation of SIRT6. Molecules 2020, 25, 1054. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compartment | Concentration Range of T1AM | Method | Reference | |
|---|---|---|---|---|
| Human and rat serum | 0.15–0.30 pmol/mL | HPLC MS-MS | Saba et al. 2010 [35] | |
| 14–66 pmol/mL | CLIA | Hoefig et al. 2011 [39] | ||
| Rat tissues | Lung | 5.61 ± 1,53 pmol/g | HPLC MS-MS | Saba et al. 2010 [35] |
| Heart | 6.60 ± 1.36 pmol/g | |||
| Stomach | 15.46 ± 6.93 pmol/g | |||
| Muscle | 25.02 ± 6.95 pmol/g | |||
| Kidney | 36.08 ± 10.42 pmol/g | |||
| Liver | 92.92 ± 28.46 pmol/g | |||
| Mouse tissues | Brain | 0.39 ± 0.102 pmol/g | HPLC MS-MS | Manni et al. 2013 [40] |
| Liver | 7.68 ± 0.85 pmol/g | Assadi-Porter et al. 2018 [41] | ||
| Adipose | 0.493 ± 0.17 pmol/g | |||
| Muscle | 19.84 ± 3.57 pmol/g | |||
| Heart | 18.15 ± 4.38 pmol/g | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rutigliano, G.; Bandini, L.; Sestito, S.; Chiellini, G. 3-Iodothyronamine and Derivatives: New Allies Against Metabolic Syndrome? Int. J. Mol. Sci. 2020, 21, 2005. https://doi.org/10.3390/ijms21062005
Rutigliano G, Bandini L, Sestito S, Chiellini G. 3-Iodothyronamine and Derivatives: New Allies Against Metabolic Syndrome? International Journal of Molecular Sciences. 2020; 21(6):2005. https://doi.org/10.3390/ijms21062005
Chicago/Turabian StyleRutigliano, Grazia, Lavinia Bandini, Simona Sestito, and Grazia Chiellini. 2020. "3-Iodothyronamine and Derivatives: New Allies Against Metabolic Syndrome?" International Journal of Molecular Sciences 21, no. 6: 2005. https://doi.org/10.3390/ijms21062005
APA StyleRutigliano, G., Bandini, L., Sestito, S., & Chiellini, G. (2020). 3-Iodothyronamine and Derivatives: New Allies Against Metabolic Syndrome? International Journal of Molecular Sciences, 21(6), 2005. https://doi.org/10.3390/ijms21062005