Molecular Mechanisms of PARP-1 Inhibitor 7-Methylguanine

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
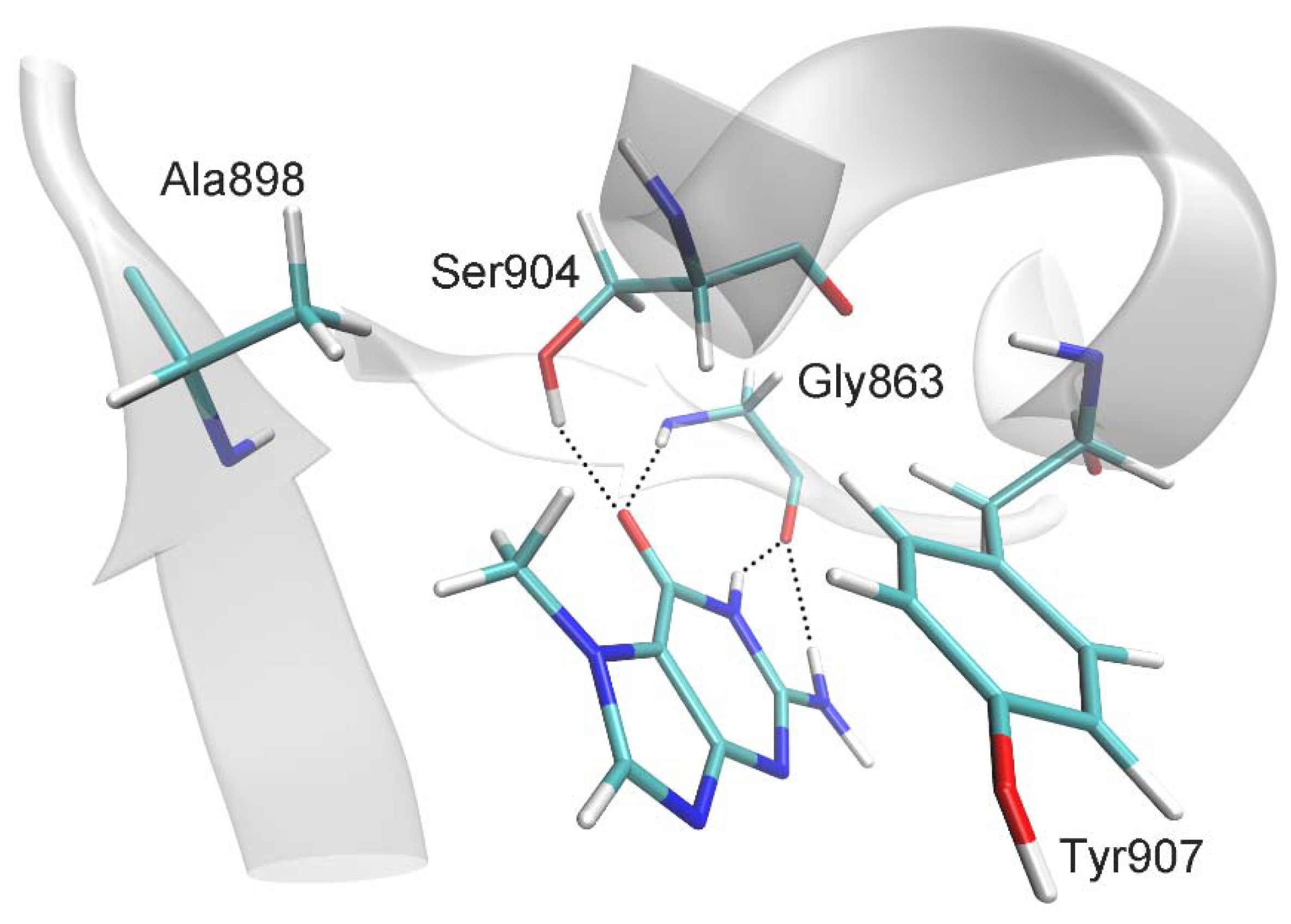
2.1. MD Modeling
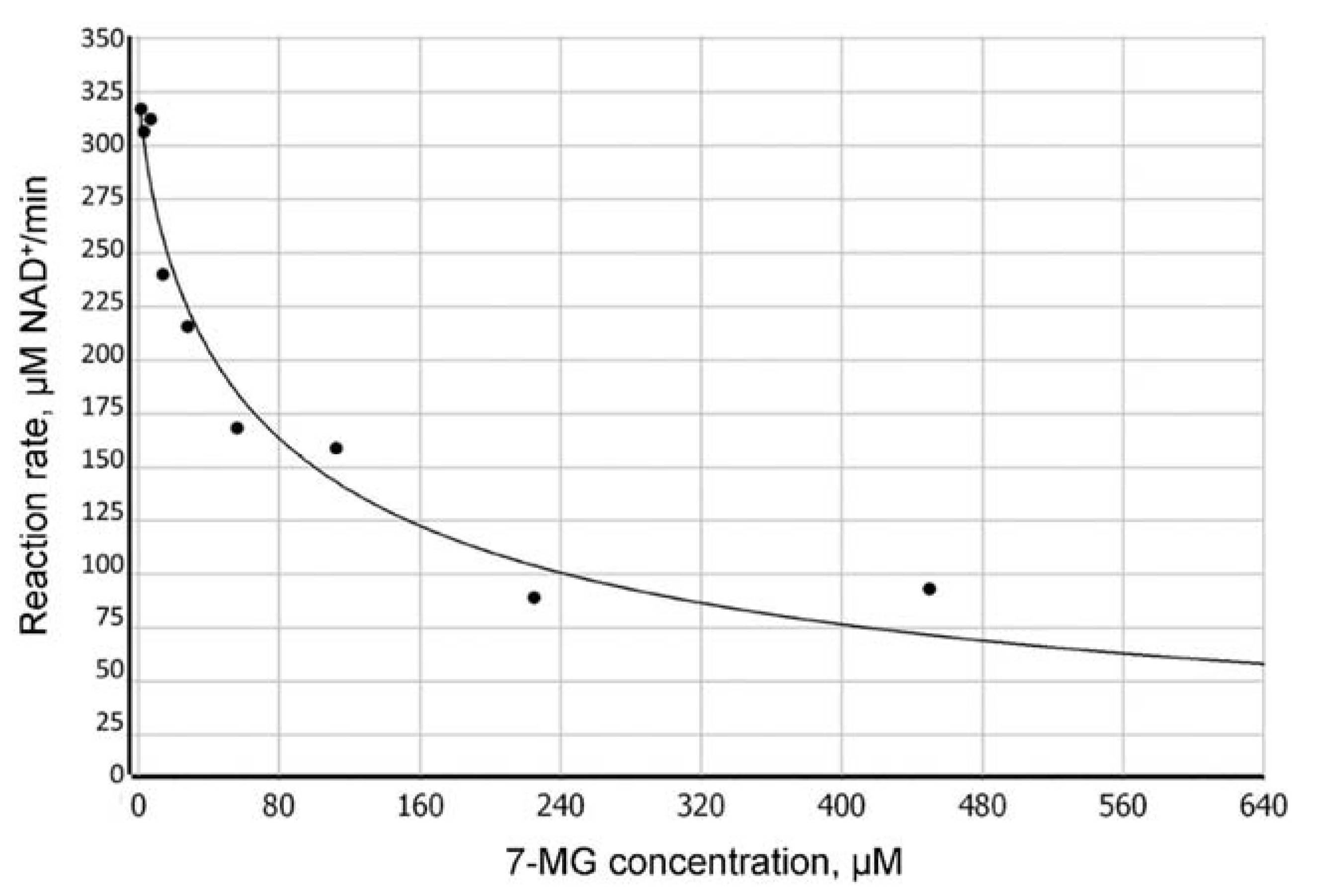
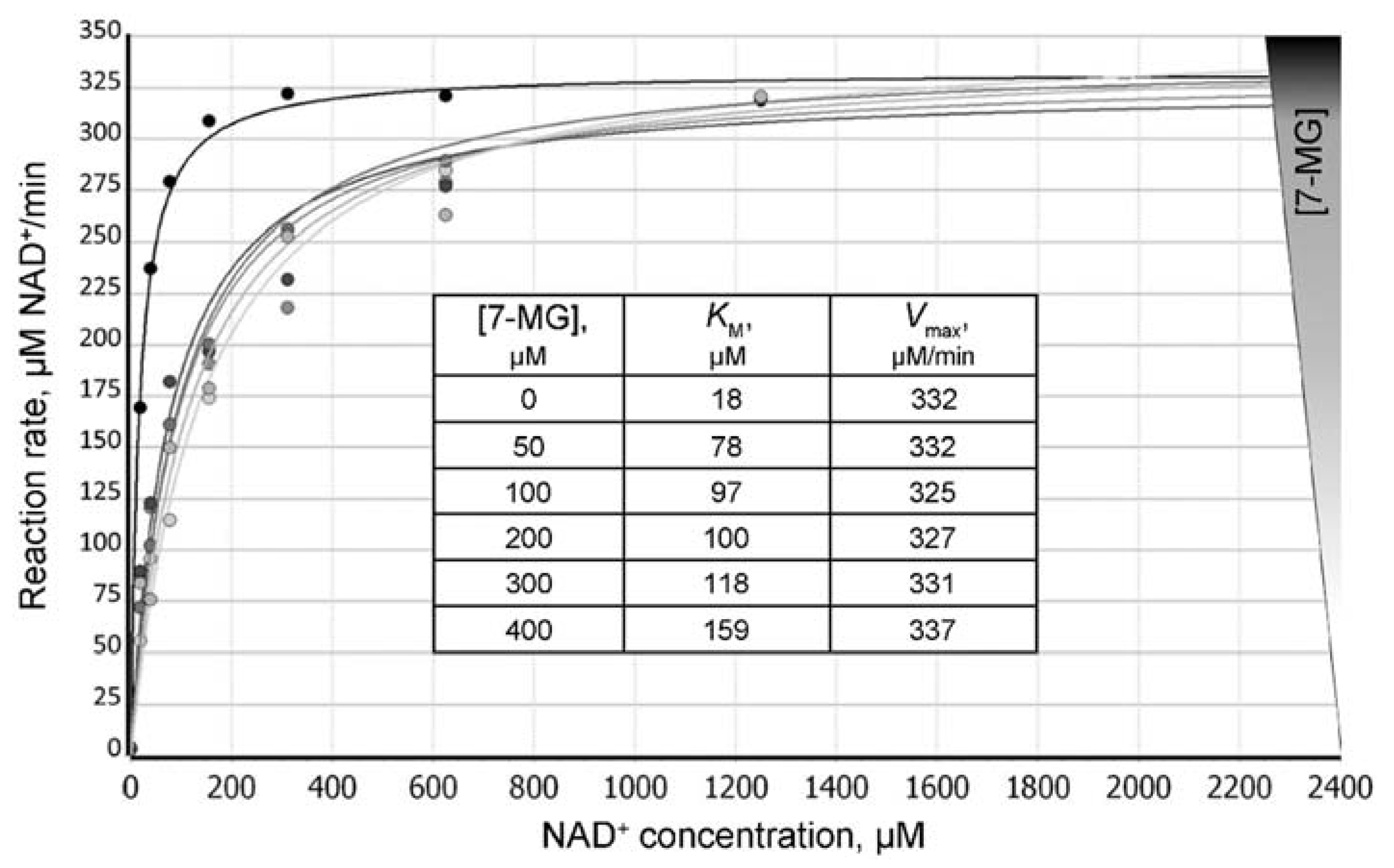
2.2. Fluorescence Anisotropy Analysis
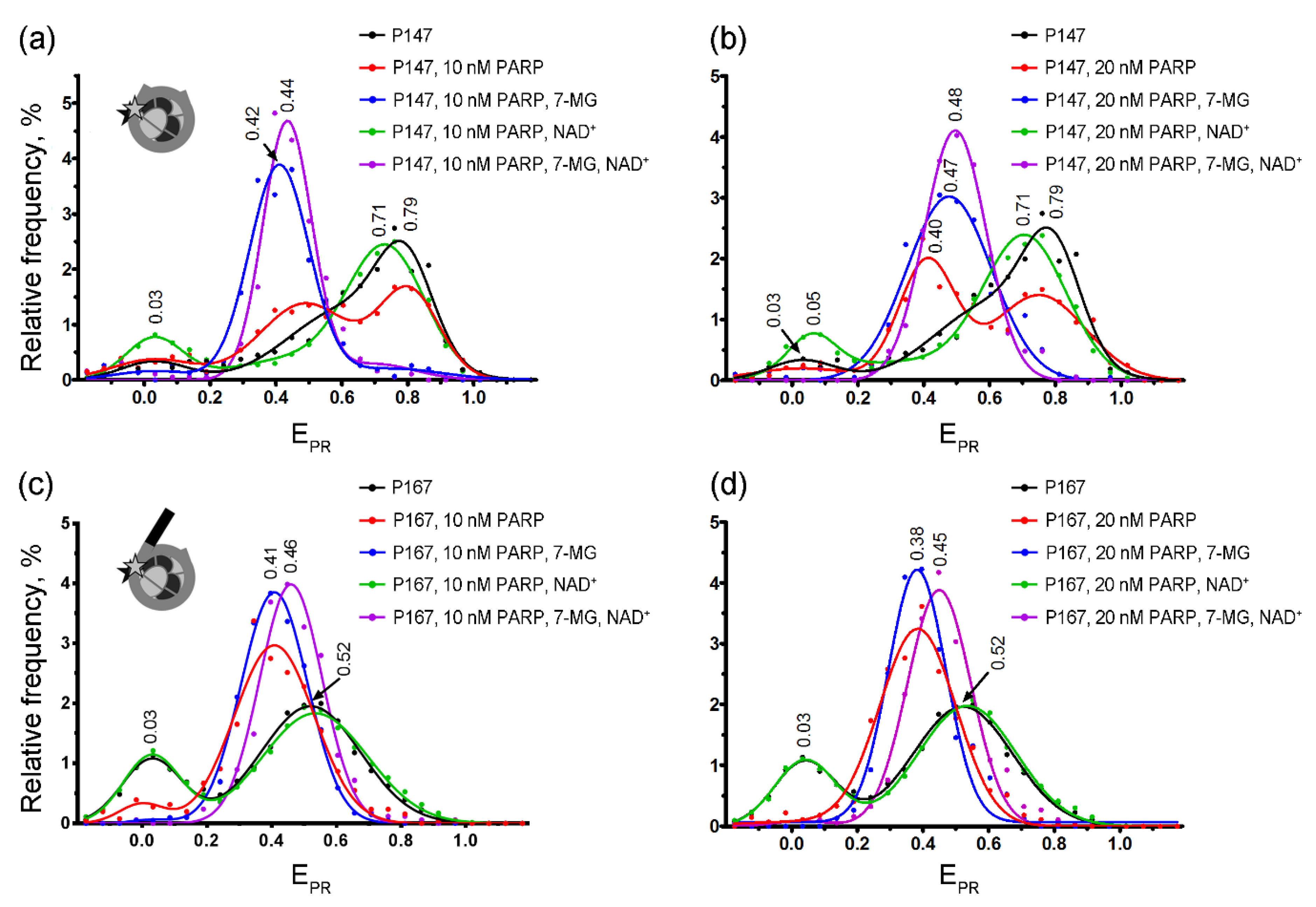
2.3. spFRET Analysis
3. Discussion
4. Materials and Methods
4.1. Molecular Modeling
4.2. Fluorescence Anisotropy Assay
4.3. spFRET Microscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PARP-1 | poly(ADP-ribose)polymerase 1 |
| PAR | poly(ADP-ribose) |
| 7-MG | 7-methylguanine |
| MD | molecular dynamics |
| spFRET | Single-particle Förster resonance energy transfer |
References
- Frampton, J.E. Olaparib: A review of its use as maintenance therapy in patients with ovarian cancer. BioDrugs 2015, 29, 143–150. [Google Scholar] [CrossRef]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP inhibitors in ovarian cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Gillard, M.; Lipkowitz, S.; Lee, J.M. Update on PARP inhibitors in breast cancer. Curr. Treat. Options Oncol. 2018, 19, 21. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [Green Version]
- Drenichev, M.S.; Mikhailov, S.N. Poly(ADP-ribose)–a unique natural polymer structural features, biological role and approaches to the chemical synthesis. Nucleosides Nucleotides Nucleic Acids 2015, 34, 258–276. [Google Scholar] [CrossRef]
- Hassler, M.; Ladurner, A.G. Towards a structural understanding of PARP1 activation and related signalling ADP-ribosyl-transferases. Curr. Opin. Struct. Biol. 2012, 22, 721–729. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodeling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Martin, S.A.; Lord, C.J.; Ashworth, A. DNA repair deficiency as a therapeutic target in cancer. Curr. Opin. Genet. Dev. 2008, 18, 80–86. [Google Scholar] [CrossRef]
- Ferraris, D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A.; Dhillon, S. Olaparib tablet: A review in ovarian cancer maintenance therapy. Target Oncol. 2018, 13, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, S.E.; Davis, C.C.; Byers, K.F. Olaparib: A novel therapy for metastatic breast cancer in patients with a BRCA1/2 mutation. J. Adv. Pract. Oncol. 2019, 10, 167–174. [Google Scholar] [PubMed]
- Jain, P.G.; Patel, B.D. Medicinal chemistry approaches of poly ADP-ribose polymerase 1 (PARP1) inhibitors as anticancer agents-a recent update. Eur. J. Med. Chem. 2019, 165, 198–215. [Google Scholar] [CrossRef]
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 inhibitors: Antitumor drug design. Acta Naturae 2015, 7, 27–37. [Google Scholar] [CrossRef]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A., Jr.; Piccart, M. An update on PARP inhibitors–moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, P.Y.; Wang, Y.T.; Yang, G.F.; Zhang, A.; Miao, Z.H. An update on poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors: Opportunities and challenges in cancer therapy. J. Med. Chem. 2016, 59, 9575–9598. [Google Scholar] [CrossRef]
- Ohmoto, A.; Yachida, S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 2017, 10, 5195–5208. [Google Scholar] [CrossRef] [Green Version]
- Geraets, L.; Moonen, H.J.; Wouters, E.F.; Bast, A.; Hageman, G.J. Caffeine metabolites are inhibitors of the nuclear enzyme poly(ADP-ribose)polymerase-1 at physiological concentrations. Biochem. Pharmacol. 2006, 72, 902–910. [Google Scholar] [CrossRef]
- Geraets, L.; Haegens, A.; Weseler, A.R.; Brauers, K.; Vernooy, J.H.; Wouters, E.F.; Bast, A.; Hageman, G.J. Inhibition of acute pulmonary and systemic inflammation by 1,7-dimethylxanthine. Eur. J. Pharmacol. 2010, 629, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Nilov, D.K.; Yashina, K.I.; Gushchina, I.V.; Zakharenko, A.L.; Sukhanova, M.V.; Lavrik, O.I.; Švedas, V.K. 2,5-Diketopiperazines: A new class of poly(ADP-ribose)polymerase inhibitors. Biochemistry (Mosc.) 2018, 83, 152–158. [Google Scholar] [CrossRef]
- Nilov, D.K.; Tararov, V.I.; Kulikov, A.V.; Zakharenko, A.L.; Gushchina, I.V.; Mikhailov, S.N.; Lavrik, O.I.; Švedas, V.K. Inhibition of poly(ADP-ribose) polymerase by nucleic acid metabolite 7-methylguanine. Acta Naturae 2016, 8, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Nilov, D.; Kirsanov, K.; Antoshina, E.; Maluchenko, N.; Feofanov, A.; Kurgina, T.; Zakharenko, A.; Khodyreva, S.; Gerasimova, N.; Studitsky, V.; et al. 7-Methylguanine: A natural DNA repair inhibitor and a promising anticancer compound. FEBS Open Bio 2018, 8. [Google Scholar] [CrossRef]
- Ruf, A.; de Murcia, G.; Schulz, G.E. Inhibitor and NAD+ binding to poly(ADP-ribose) polymerase as derived from crystal structures and homology modeling. Biochemistry 1998, 37, 3893–3900. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Szabó, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Kurgina, T.A.; Anarbaev, R.O.; Sukhanova, M.V.; Lavrik, O.I. A rapid fluorescent method for the real-time measurement of poly(ADP-ribose) polymerase 1 activity. Anal. Biochem. 2018, 545, 91–97. [Google Scholar] [CrossRef]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Valieva, M.E.; Armeev, G.A.; Kudryashova, K.S.; Gerasimova, N.S.; Shaytan, A.K.; Kulaeva, O.I.; McCullough, L.L.; Formosa, T.; Georgiev, P.G.; Kirpichnikov, M.P.; et al. Large-scale ATP-independent nucleosome unfolding by a histone chaperone. Nat. Struct. Mol. Biol. 2016, 23, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Sultanov, D.C.; Gerasimova, N.S.; Kudryashova, K.S.; Maluchenko, N.V.; Kotova, E.Y.; Langelier, M.F.; Pascal, J.M.; Kirpichnikov, M.P.; Feofanov, A.V.; Studitsky, V.M. Unfolding of core nucleosomes by PARP-1 revealed by spFRET microscopy. AIMS Genet. 2017, 4, 21–31. [Google Scholar] [CrossRef]
- Buning, R.; van Noort, J. Single-pair FRET experiments on nucleosome conformational dynamics. Biochimie 2010, 92, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, K.S.; Chertkov, O.V.; Nikitin, D.V.; Pestov, N.A.; Kulaeva, O.I.; Efremenko, A.V.; Solonin, A.S.; Kirpichnikov, M.P.; Studitsky, V.M.; Feofanov, A.V. Preparation of mononucleosomal templates for analysis of transcription with RNA polymerase using spFRET. Methods Mol. Biol. 2015, 1288, 395–412. [Google Scholar] [PubMed] [Green Version]
- Lyubitelev, A.V.; Kudryashova, K.S.; Mikhaylova, M.S.; Malyuchenko, N.V.; Chertkov, O.V.; Studitsky, V.M.; Feofanov, A.V.; Kirpichnikov, M.P. Change in linker DNA conformation upon histone H1.5 binding to nucleosome: Fluorescent microscopy of single complexes. Moscow Univ. Biol. Sci. Bull. 2015, 71, 108–113. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping poly(ADP-ribose) polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Gerasimova, N.S.; Valieva, M.E.; Sultanov, D.C.; Kudryashova, K.S.; Maluchenko, N.V.; Kotova, E.S.; Kirpichnikov, M.P.; Studitsky, V.M.; Feofanov, A.V. Complexes of nucleosomal nanoparticles with proteins: spFRET microscopy study of olaparib and PARP-1 binding to core nucleosomes. In Microscopy and Imaging Science: Practical Approaches to Applied Research and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2017; pp. 55–61. [Google Scholar]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., 3rd; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015; pp. 1–883. [Google Scholar]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead finder: An approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Sukhanova, M.V.; Khodyreva, S.N.; Novikov, F.N.; Stroylov, V.S.; Nilov, D.K.; Chilov, G.G.; Švedas, V.K.; Lavrik, O.I. Improved procedure of the search for poly(ADP-Ribose) polymerase-1 potential inhibitors with the use of the molecular docking approach. Mol. Biol. (Mosc.) 2011, 45, 517–521. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Amé, J.C.; Kalisch, T.; Dantzer, F.; Schreiber, V. Purification of recombinant poly(ADP-ribose) polymerases. Methods Mol. Biol. 2011, 780, 135–152. [Google Scholar]
- Kulaeva, O.I.; Gaykalova, D.A.; Pestov, N.A.; Golovastov, V.V.; Vassylyev, D.G.; Artsimovitch, I.; Studitsky, V.M. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat. Struct. Mol. Biol. 2009, 16, 1272–1278. [Google Scholar] [CrossRef] [Green Version]
- Gaykalova, D.A.; Kulaeva, O.I.; Volokh, O.; Shaytan, A.K.; Hsieh, F.K.; Kirpichnikov, M.P.; Sokolova, O.S.; Studitsky, V.M. Structural analysis of nucleosomal barrier to transcription. Proc. Natl. Acad. Sci. USA 2015, 112, E5787–E5795. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, D.; Chua, E.Y.D.; Davey, C.A. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol. 2010, 403, 1–10. [Google Scholar] [CrossRef]
- Morozov, A.V.; Fortney, K.; Gaykalova, D.A.; Studitsky, V.M.; Widom, J.; Siggia, E.D. Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res. 2009, 37, 4707–4722. [Google Scholar] [CrossRef]
- Ausio, J.; Dong, F.; van Holde, K.E. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone “tails” in the stabilization of the nucleosome. J. Mol. Biol. 1989, 206, 451–463. [Google Scholar] [CrossRef]
- Kornberg, R.D.; LaPointe, J.W.; Lorch, Y. Preparation of nucleosomes and chromatin. Methods Enzymol. 1989, 170, 3–14. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance, Å | |
| 7-MG:CO:O ··· Gly863:H | 2.0 ± 0.2 |
| 7-MG:CO:O ··· Ser904:OG:HG | 2.5 ± 0.7 |
| 7-MG:NH:H ··· Gly863:O | 1.9 ± 0.1 |
| 7-MG:NH2:H ··· Gly863:O | 2.4 ± 0.3 |
| 7-MG:CH3:C ··· Ala898:CB | 4.0 ± 0.4 |
| C(7-MG fused rings) ··· C(Tyr907 benzene ring) 1 | 3.6 ± 0.2 |
| Angle, deg | |
| 7-MG:CO:O ··· Gly863:H ··· Gly863:N | 162 ± 11 |
| 7-MG:CO:O ··· Ser904:OG:HG ··· Ser904:OG | 134 ± 34 |
| 7-MG:NH:N ··· 7-MG:NH:H ··· Gly863:O | 153 ± 11 |
| 7-MG:NH2:N ··· 7-MG:NH2:H ··· Gly863:O | 138 ± 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilov, D.; Maluchenko, N.; Kurgina, T.; Pushkarev, S.; Lys, A.; Kutuzov, M.; Gerasimova, N.; Feofanov, A.; Švedas, V.; Lavrik, O.; et al. Molecular Mechanisms of PARP-1 Inhibitor 7-Methylguanine. Int. J. Mol. Sci. 2020, 21, 2159. https://doi.org/10.3390/ijms21062159
Nilov D, Maluchenko N, Kurgina T, Pushkarev S, Lys A, Kutuzov M, Gerasimova N, Feofanov A, Švedas V, Lavrik O, et al. Molecular Mechanisms of PARP-1 Inhibitor 7-Methylguanine. International Journal of Molecular Sciences. 2020; 21(6):2159. https://doi.org/10.3390/ijms21062159
Chicago/Turabian StyleNilov, Dmitry, Natalya Maluchenko, Tatyana Kurgina, Sergey Pushkarev, Alexandra Lys, Mikhail Kutuzov, Nadezhda Gerasimova, Alexey Feofanov, Vytas Švedas, Olga Lavrik, and et al. 2020. "Molecular Mechanisms of PARP-1 Inhibitor 7-Methylguanine" International Journal of Molecular Sciences 21, no. 6: 2159. https://doi.org/10.3390/ijms21062159
APA StyleNilov, D., Maluchenko, N., Kurgina, T., Pushkarev, S., Lys, A., Kutuzov, M., Gerasimova, N., Feofanov, A., Švedas, V., Lavrik, O., & Studitsky, V. M. (2020). Molecular Mechanisms of PARP-1 Inhibitor 7-Methylguanine. International Journal of Molecular Sciences, 21(6), 2159. https://doi.org/10.3390/ijms21062159