Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes


Abstract
:
1. Introduction
2. Results
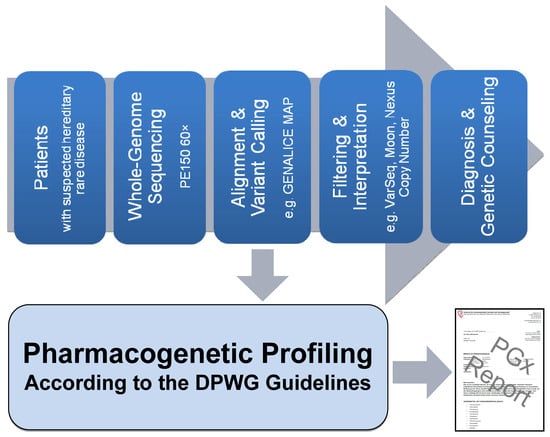
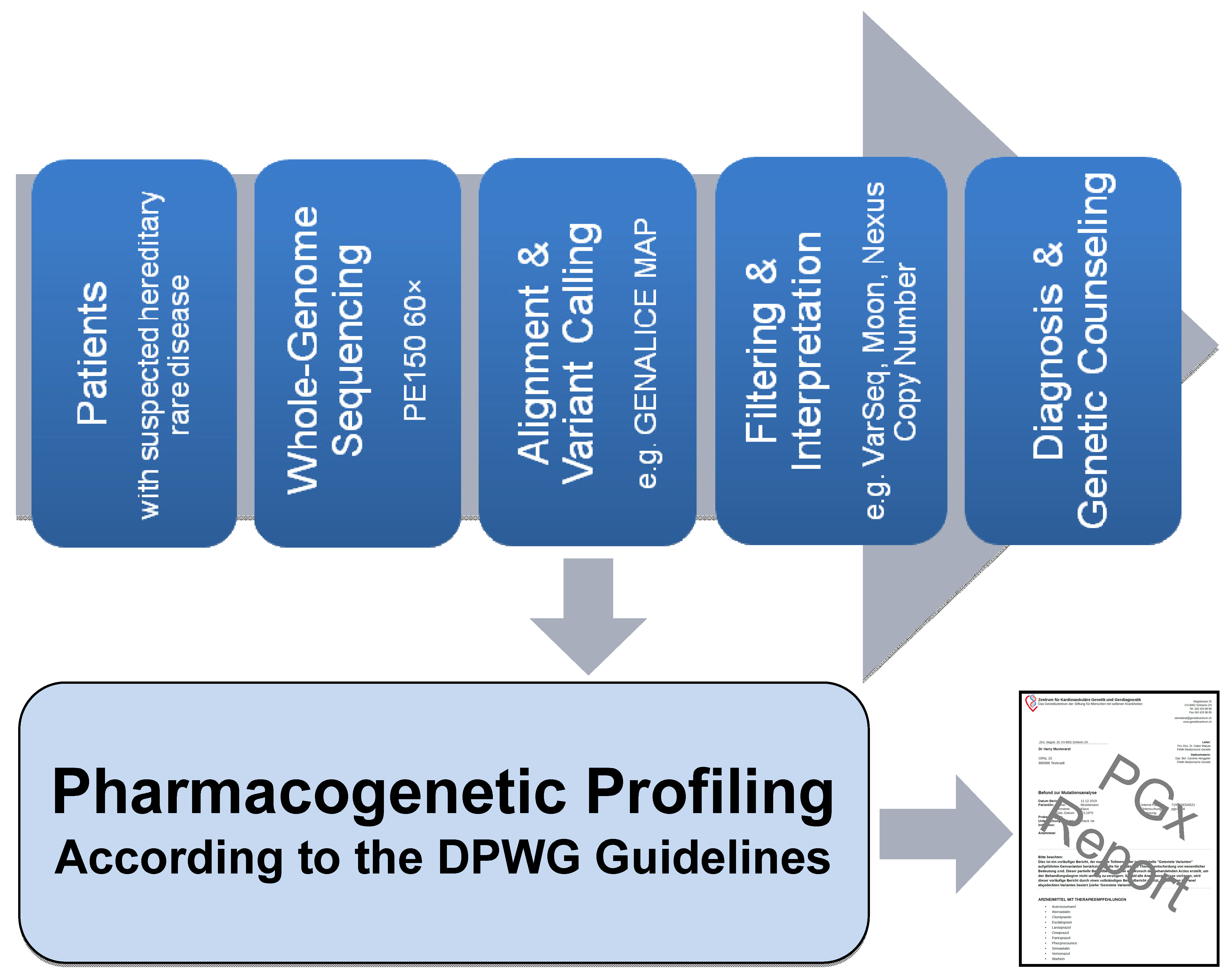
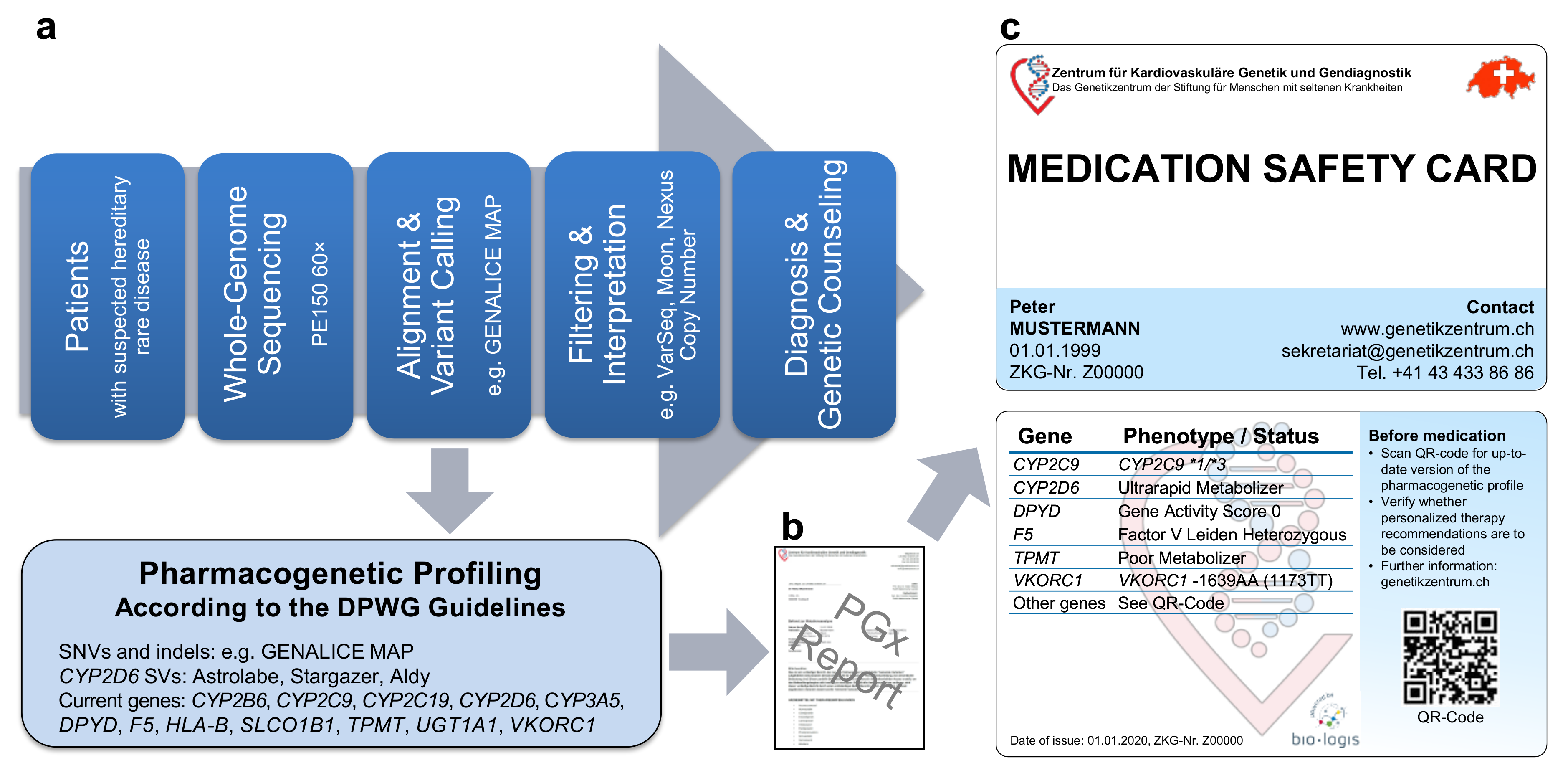
2.1. Pipeline from Raw Data to Molecular Genetic Diagnosis, PGx Report, and Medication Safety Card (MSC)
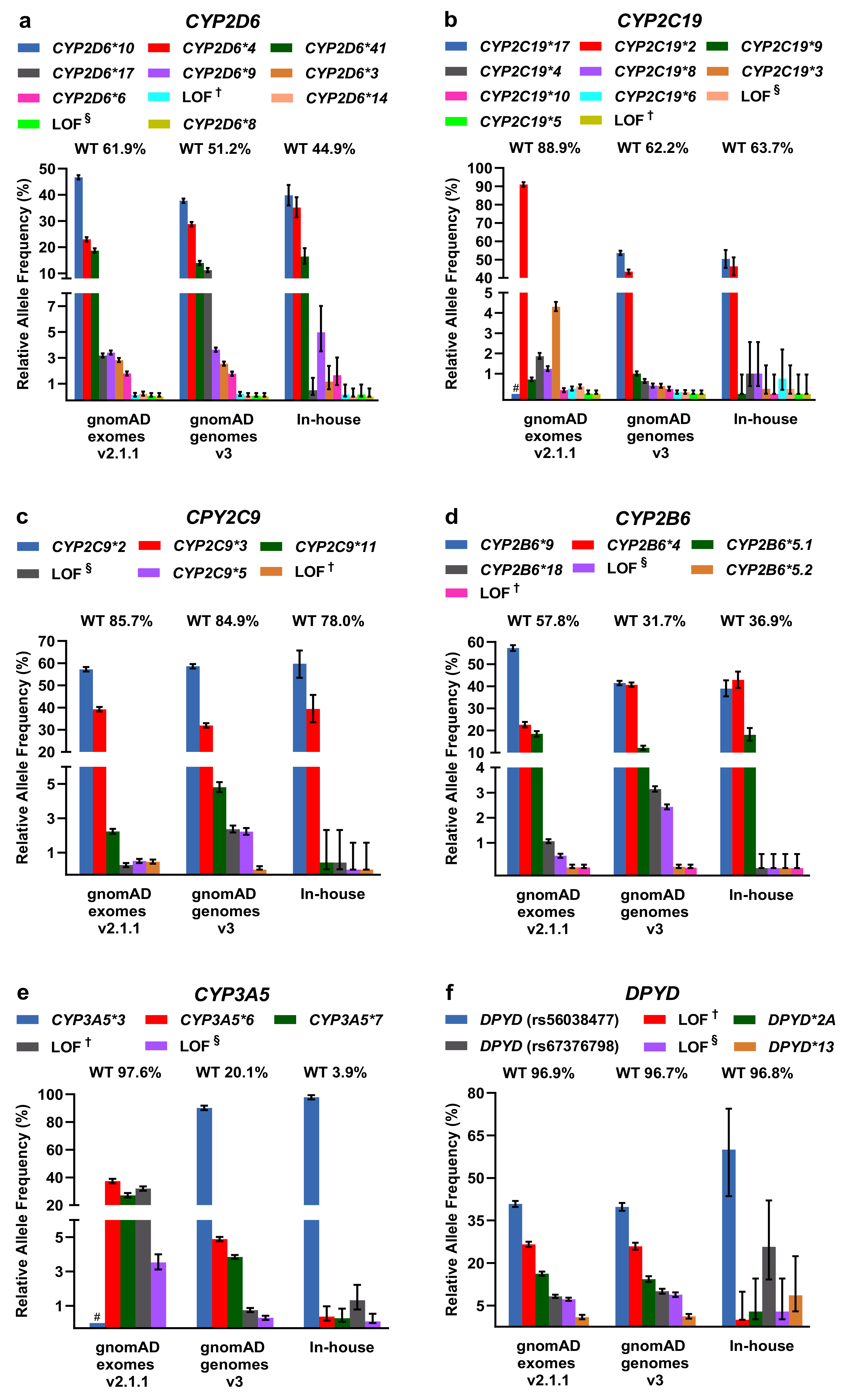
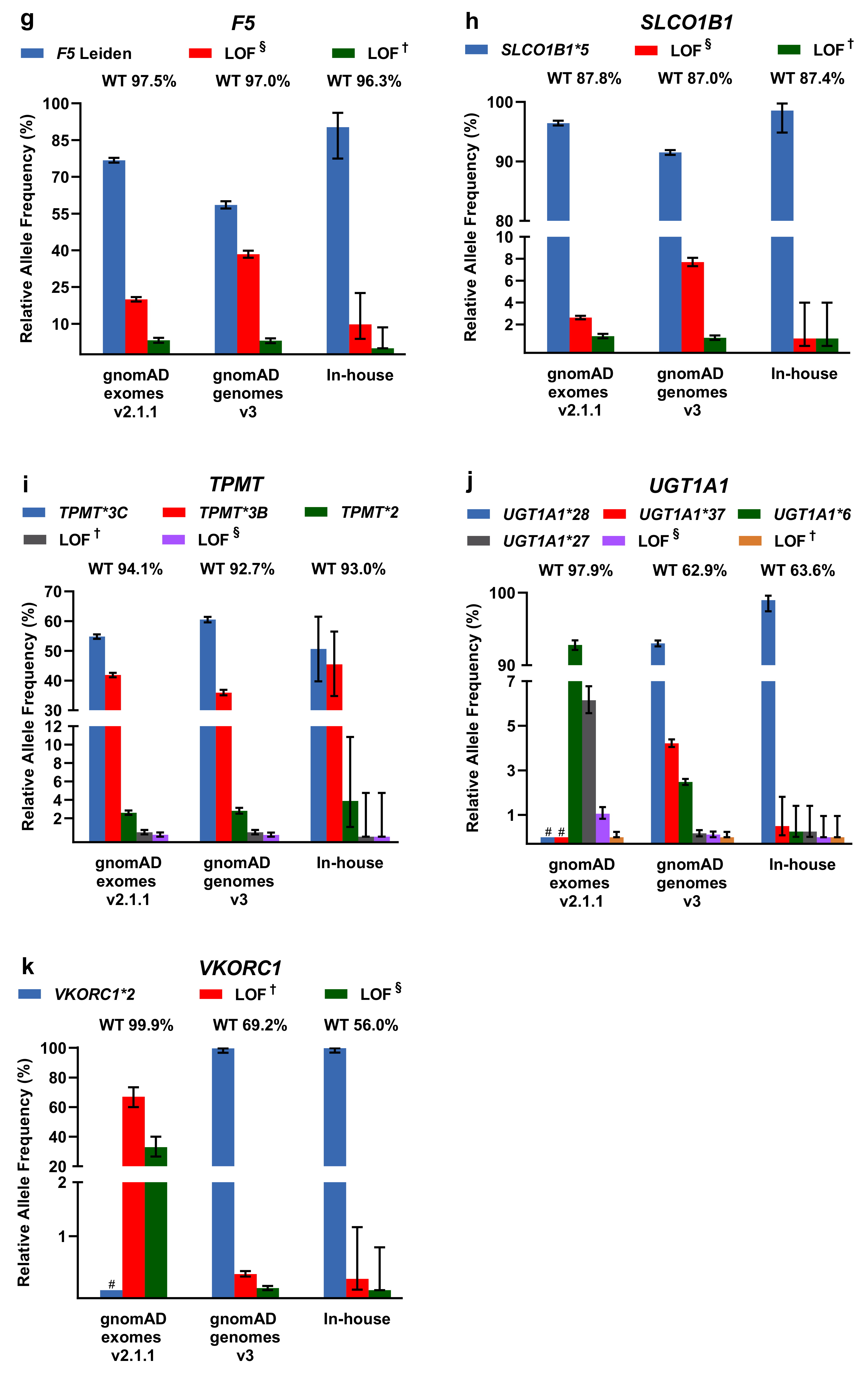
2.2. Evaluation of PGx Variants in gnomAD and Our In-House Cohort
2.3. Comparison of CYP2D6 Calling Tools from WGS Data
3. Discussion
4. Materials and Methods
4.1. PGx Profiling from WGS Data as well as Comparison of WGS and WES for PGx Profiling
4.2. Analysis of Star Alleles and Loss-of-Function Variants in gnomAD and in Our In-House WGS Cohort
4.3. Evaluation of CYP2D6 Variant Callers
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Strengthening Pharmacovigilance to Reduce Adverse Effects of Medicines. Available online: Europa.eu/rapid/press-release_MEMO-08-782_de.htm?locale=en (accessed on 24 January 2020).
- Watanabe, J.H.; McInnis, T.; Hirsch, J.D. Cost of Prescription Drug–Related Morbidity and Mortality. Ann. Pharmacother. 2018, 52, 829–837. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mechanic, L.E.; Long, R.; Freedman, A.N. Pharmacogenomics in oncology care. Front. Genet. 2014, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.S.; Fulton, R.S.; Qin, X.; Mardis, E.R.; Nickerson, D.A.; Scherer, S. PGRNseq: A targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet. Genom. 2016, 26, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte--an update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- van der Wouden, C.H.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; Dávila-Fajardo, C.L.; Deneer, V.H.; Dolžan, V.; Ingelman-Sundberg, M.; Jönsson, S.; Karlsson, M.O.; et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2017, 101, 341–358. [Google Scholar] [CrossRef]
- Blagec, K.; Koopmann, R.; Crommentuijn-van Rhenen, M.; Holsappel, I.; van der Wouden, C.H.; Konta, L.; Xu, H.; Steinberger, D.; Just, E.; Swen, J.J.; et al. Implementing pharmacogenomics decision support across seven European countries: The Ubiquitous Pharmacogenomics (U-PGx) project. J. Am. Med. Inform. Assoc. 2018, 25, 893–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, O.; Scott, S.A.; Ellis, S.B.; Overby, C.L.; Ludtke, A.; Hulot, J.-S.; Hall, J.; Chatani, K.; Myers, K.; Kannry, J.L.; et al. The CLIPMERGE PGx Program: Clinical implementation of personalized medicine through electronic health records and genomics-pharmacogenomics. Clin. Pharmacol. Ther. 2013, 94, 214–217. [Google Scholar] [CrossRef]
- Rasmussen-Torvik, L.J.; Stallings, S.C.; Gordon, A.S.; Almoguera, B.; Basford, M.A.; Bielinski, S.J.; Brautbar, A.; Brilliant, M.H.; Carrell, D.S.; Connolly, J.J.; et al. Design and anticipated outcomes of the eMERGE-PGx project: A multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin. Pharmacol. Ther. 2014, 96, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspar, S.M.; Dubacher, N.; Kopps, A.M.; Meienberg, J.; Henggeler, C.; Matyas, G. Clinical sequencing: From raw data to diagnosis with lifetime value. Clin. Genet. 2018, 93, 508–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twist, G.P.; Gaedigk, A.; Miller, N.A.; Farrow, E.G.; Willig, L.K.; Dinwiddie, D.L.; Petrikin, J.E.; Soden, S.E.; Herd, S.; Gibson, M.; et al. Constellation: A tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom. Med. 2016, 1, 15007. [Google Scholar] [CrossRef] [PubMed]
- Numanagić, I.; Malikić, S.; Ford, M.; Qin, X.; Toji, L.; Radovich, M.; Skaar, T.C.; Pratt, V.M.; Berger, B.; Scherer, S.; et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat. Commun. 2018, 9, 828. [Google Scholar] [CrossRef]
- Lee, S.B.; Wheeler, M.M.; Thummel, K.E.; Nickerson, D.A. Calling Star Alleles With Stargazer in 28 Pharmacogenes With Whole Genome Sequences. Clin. Pharmacol. Ther. 2019, 106, 1328–1337. [Google Scholar] [CrossRef] [Green Version]
- Tremmel, R.; Klein, K.; Battke, F.; Fehr, S.; Winter, S.; Scheurenbrand, T.; Schaeffeler, E.; Biskup, S.; Schwab, M.; Zanger, U.M. Copy number variation profiling in pharmacogenes using panel-based exome resequencing and correlation to human liver expression. Hum. Genet. 2020, 139, 137–149. [Google Scholar] [CrossRef]
- Reisberg, S.; Krebs, K.; Lepamets, M.; Kals, M.; Mägi, R.; Metsalu, K.; Lauschke, V.M.; Vilo, J.; Milani, L. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: Challenges and solutions. Genet. Med. 2019, 21, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- VKORC1-Genotypisierung. Available online: Charite.de/fileadmin/user_upload/microsites/m_cc05/ilp/referenzdb/20204037.htm (accessed on 24 January 2020).
- Gaedigk, A.; Turner, A.; Everts, R.E.; Scott, S.A.; Aggarwal, P.; Broeckel, U.; McMillin, G.A.; Melis, R.; Boone, E.C.; Pratt, V.M.; et al. Characterization of Reference Materials for Genetic Testing of CYP2D6 Alleles: A GeT-RM Collaborative Project. J. Mol. Diagn. 2019, 21, 1034–1052. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gage, B.F.; Bass, A.R.; Lin, H.; Woller, S.C.; Stevens, S.M.; Al-Hammadi, N.; Li, J.; Rodríguez, T.; Miller, J.P.; McMillin, G.A.; et al. Effect of Genotype-Guided Warfarin Dosing on Clinical Events and Anticoagulation Control Among Patients Undergoing Hip or Knee Arthroplasty: The GIFT Randomized Clinical Trial. JAMA 2017, 318, 1115. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin. Pharmacokinet. 2009, 48, 689–723. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Delaneau, O.; Coulonges, C.; Zagury, J.-F. Shape-IT: New rapid and accurate algorithm for haplotype inference. BMC Bioinformatics 2008, 9, 540. [Google Scholar] [CrossRef] [Green Version]
- Qiao, W.; Yang, Y.; Sebra, R.; Mendiratta, G.; Gaedigk, A.; Desnick, R.J.; Scott, S.A. Long-Read Single Molecule Real-Time Full Gene Sequencing of Cytochrome P450-2D6. Hum. Mutat. 2016, 37, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Buermans, H.P.J.; Vossen, R.H.A.M.; Anvar, S.Y.; Allard, W.G.; Guchelaar, H.-J.; White, S.J.; den Dunnen, J.T.; Swen, J.J.; van der Straaten, T. Flexible and Scalable Full-Length CYP2D6 Long Amplicon PacBio Sequencing. Hum. Mutat. 2017, 38, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Liau, Y.; Maggo, S.; Miller, A.L.; Pearson, J.F.; Kennedy, M.A.; Cree, S.L. Nanopore sequencing of the pharmacogene CYP2D6 allows simultaneous haplotyping and detection of duplications. Pharmacogenomics 2019, 20, 1033–1047. [Google Scholar] [CrossRef]
- Van der Wouden, C.H.; van Rhenen, M.H.; Jama, W.O.M.; Ingelman-Sundberg, M.; Lauschke, V.M.; Konta, L.; Schwab, M.; Swen, J.J.; Guchelaar, H.-J. Development of the PGx-Passport: A Panel of Actionable Germline Genetic Variants for Pre-Emptive Pharmacogenetic Testing. Clin. Pharmacol. Ther. 2019, 106, 866–873. [Google Scholar] [CrossRef] [PubMed]
- The G-Standaard: The Medicines Standard in Healthcare. Available online: Knmp.nl/producten/gebruiksrecht-g-standaard/informatie-over-de-g-standaard/the-g-standaard-the-medicines-standard-in-healthcare (accessed on 10 January 2020).
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Selamet Tierney, E.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus Losartan in Children and Young Adults with Marfan’s Syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, D.A.; Gehr, T.W.B.; Ghosh, S. Clinical pharmacokinetics of losartan. Clin. Pharmacokinet. 2005, 44, 797–814. [Google Scholar] [CrossRef]
- Plüss, M.; Kopps, A.M.; Keller, I.; Meienberg, J.; Caspar, S.M.; Dubacher, N.; Bruggmann, R.; Vogel, M.; Matyas, G. Need for speed in accurate whole-genome data analysis: GENALICE MAP challenges BWA/GATK more than PEMapper/PECaller and Isaac. Proc. Natl. Acad. Sci. USA 2017, 114, E8320–E8322. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–33. [Google Scholar] [CrossRef]
- Meienberg, M.; Zerjavic, K.; Keller, I.; Okoniewski, M.; Patrignani, A.; Ludin, K.; Xu, Z.; Steinmann, B.; Röthlisberger, B.; Schlapbach, R.; et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 2015, 43, e45. [Google Scholar] [CrossRef] [Green Version]
- Colombo, S.; Rauch, A.; Rotger, M.; Fellay, J.; Martinez, R.; Fux, C.; Thurnheer, C.; Günthard, H.F.; Goldstein, D.B.; Furrer, H.; et al. The HCP5 Single-Nucleotide Polymorphism: A Simple Screening Tool for Prediction of Hypersensitivity Reaction to Abacavir. J. Infect. Dis. 2008, 198, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Estellé, J.; Marco Sola, S.; Knowles, D.G.; Raineri, E.; Guigó, R.; Ribeca, P. Fast computation and applications of genome mappability. PLoS ONE 2012, 7, e30377. [Google Scholar] [CrossRef]
- Najafi, A.; Caspar, S.M.; Meienberg, J.; Rohrbach, M.; Steinmann, B.; Matyas, G. Variant filtering, digenic variants, and other challenges in clinical sequencing: A lesson from fibrillinopathies. Clin. Genet. 2020, 97, 235–245. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The Pharmacogenomics Knowledge Base. Methods Mol. Biol. 2013, 1015, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, V.M.; Everts, R.E.; Aggarwal, P.; Beyer, B.N.; Broeckel, U.; Epstein-Baak, R.; Hujsak, P.; Kornreich, R.; Liao, J.; Lorier, R.; et al. Characterization of 137 Genomic DNA Reference Materials for 28 Pharmacogenetic Genes: A GeT-RM Collaborative Project. J. Mol. Diagn. 2016, 18, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Raczy, C.; Petrovski, R.; Saunders, C.T.; Chorny, I.; Kruglyak, S.; Margulies, E.H.; Chuang, H.-Y.; Källberg, M.; Kumar, S.A.; Liao, A.; et al. Isaac: Ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 2013, 29, 2041–2043. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | gnomAD Exomes v2.1.1 | gnomAD Genomes v3 | In-House WGS Cohort | |||
|---|---|---|---|---|---|---|
| Cohort Size | 125’748 Exomes | 71’702 Genomes | 547 Genomes | |||
| Novel LOF/missense variants (not in ClinVar/HGMD/PharmGKB) 1 | n | % | n | % | n | % |
| Total variants (alleles) | 823 (4′214) | 100.0 | 512 (6′940) | 100.0 | 10 (10) | 100.0 |
| MAF > 5% | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| 0.1% < MAF < 5% | 2 | 0.2 | 8 | 1.6 | 0 | 0.0 |
| MAF < 0.1% | 821 | 99.8 | 504 | 98.4 | 10 | 100.0 |
| Known LOF/missense variants (in ClinVar/HGMD/PharmGKB) 1 | n | % | n | % | n | % |
| Total variants (alleles) | 103 (5′584) | 100.0 | 80 (2′706) | 100.0 | 4 (17) | 100.0 |
| MAF > 5% | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| 0.1% < MAF < 5% | 5 | 4.9 | 3 | 3.7 | 1 | 25.0 |
| MAF < 0.1% | 98 | 95.1 | 77 | 96.3 | 3 | 75.0 |
| DPWG variants | n | % | n | % | n | % |
| Total variants (alleles) | 40 (375′331) | 100.0 | 45 (487′758) | 100.0 | 37 (4′162) | 100.0 |
| MAF > 5% | 10 | 25.0 | 14 | 31.1 | 14 | 37.8 |
| 0.1% < MAF < 5% | 22 | 55.0 | 23 | 51.1 | 18 | 48.7 |
| MAF < 0.1% | 8 | 20.0 | 8 | 17.8 | 5 | 13.5 |
| Reference Samples | GeT-RM Consensus Genotype 2019 | Astrolabe v0.8.6.1 | Aldy v2.2.3 | Stargazer 1 v1.0.7 |
|---|---|---|---|---|
| HG00436 | *2×2/*71 | *2/*71 | *2×2/*71 | *1/*83+*2 |
| NA07029 | *1/*35 | *1/*35 | *1/*35 | *1/*35 |
| NA18959 | *2/*36+*10 | *2/*10 | *2/*36+*10 | *2/*36+*10 |
| NA19109 | *2×2/*29 | *2/*29 | *2×2/*29 | *29/*83+*2 |
| NA21781 | *2×2/*68+*4 | *2/*4 2 | *2×2/*68+*4 | *4N+*4/*68+*4 |
| NA12878 in-house | *3/(*68)+*4 | *3/*4 3 | *3/*68+*4 | *3/*4 |
| NA12873 | *1/*5 | *1/*5 | *5/*61 4 | *1/*5 |
| NA18861 | *5/*29 | *5/*29 | *5/*29 | *13C/*29 |
| HG00589 | *1/*21 | *1/*2 | *1/*21 | *1/*2 |
| NA19917 | *1/*40 | *1/*40 | *1/*40 | *1/*40 |
| NA07019 | *1/*4 | *1/*4 | *1/*4 | *1/*4 |
| NA12717 | *1/*1 | *1/*1 | *1/*1 | *1/*1 |
| HG00276 | *4/*5 | *4/*4 | *4/*5 | *4/*5 |
| NA18524 | *1/*36×2+*10 | *1/*10 | *1+*36/*36+*10 5 | *1/*10×3 |
| NA18540 | (*36+)*10/*41 | *41/*10 3 | *36+10/*61+*69 | *10×2/*41×2 |
| NA12892 | *2/*3 | *2/*3 | *2/*3 | *2/*3 |
| NA07348 | *1/*6 | *1/*6 | *1/*6 | *1/*6 |
| NA18519 | *1/*29 6 | *1/*29 | *106/*29 | *106/*29 |
| NA18966 | *1/*2 | *1/*2 | *1/*2 | *1/*2 |
| NA18992 | *1/*5 | *1/*5 | *1/*5 | *1/*13C |
| NA19226 | *2/*2×2 | *2/*2 2 | *2/*2×2 | *2/*83+*2 |
| Total true | 12 | 19 | 11 | |
| Total false | 9 | 2 | 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caspar, S.M.; Schneider, T.; Meienberg, J.; Matyas, G. Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes. Int. J. Mol. Sci. 2020, 21, 2308. https://doi.org/10.3390/ijms21072308
Caspar SM, Schneider T, Meienberg J, Matyas G. Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes. International Journal of Molecular Sciences. 2020; 21(7):2308. https://doi.org/10.3390/ijms21072308
Chicago/Turabian StyleCaspar, Sylvan M., Timo Schneider, Janine Meienberg, and Gabor Matyas. 2020. "Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes" International Journal of Molecular Sciences 21, no. 7: 2308. https://doi.org/10.3390/ijms21072308
APA StyleCaspar, S. M., Schneider, T., Meienberg, J., & Matyas, G. (2020). Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes. International Journal of Molecular Sciences, 21(7), 2308. https://doi.org/10.3390/ijms21072308