TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CFTR-Independent Approaches to Restoring Airway Mucosal Hydration
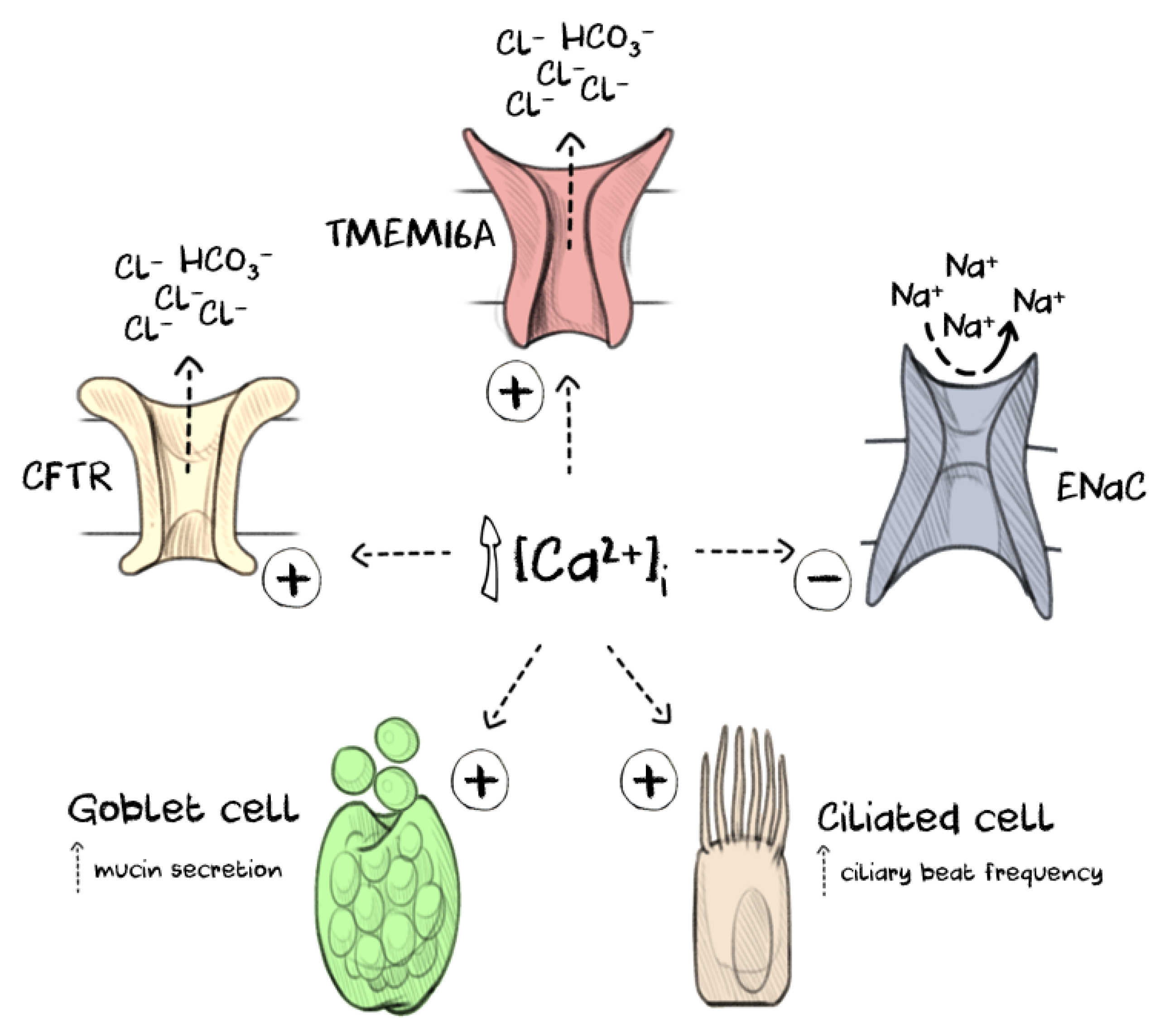
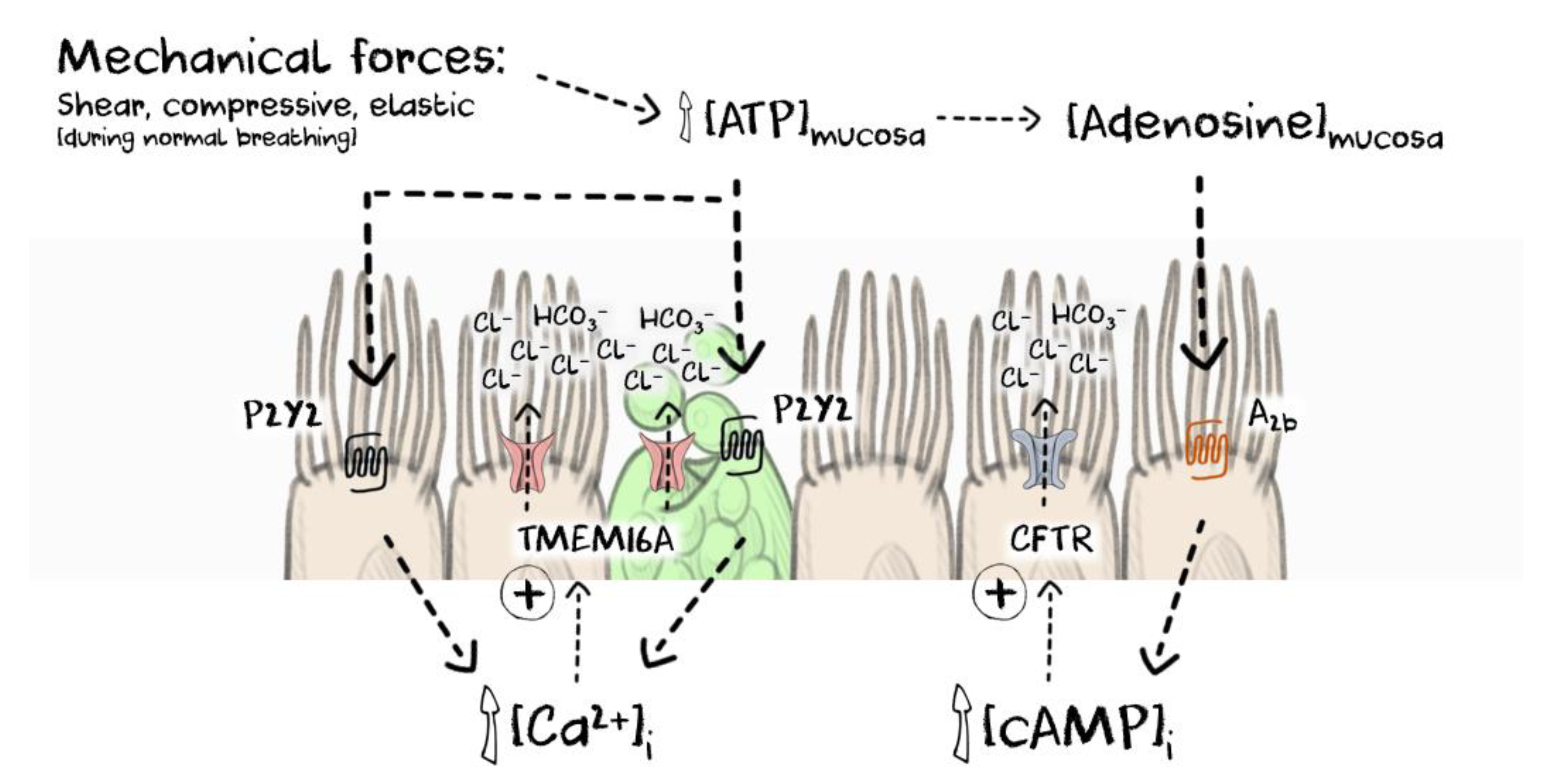
3. TMEM16A: Regulation of CaCC Function in the Airway Epithelium
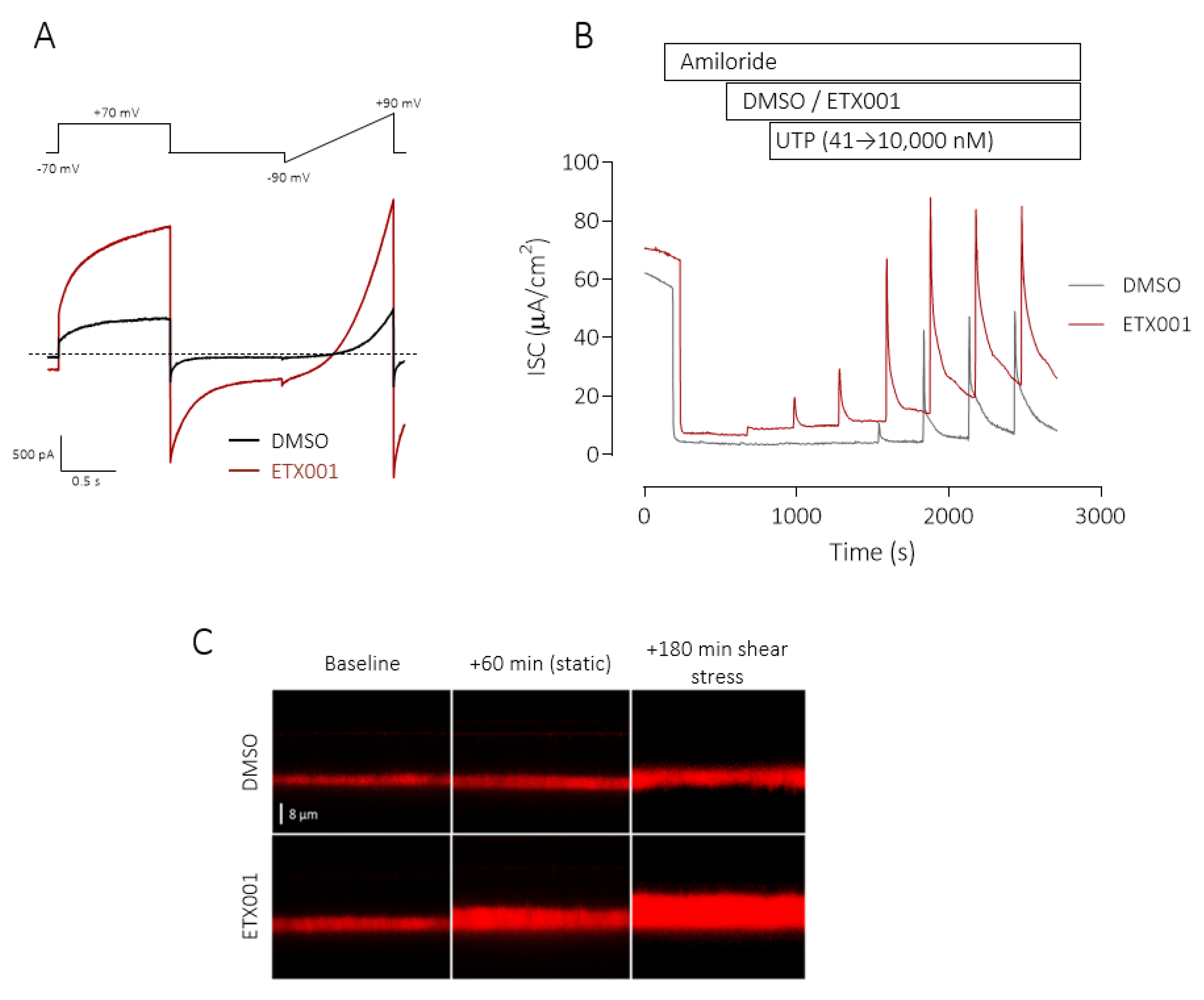
4. The Discovery and Validation of TMEM16A Potentiators
5. Additional Roles for TMEM16A in the Airways: Could TMEM16A Inhibitors be a Therapeutic Approach?
6. TMEM16A Function Outside of the Airways
7. Summary and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 1–19. [Google Scholar] [CrossRef]
- Boucher, R.C. Airway surface dehydration in cystic fibrosis: Pathogenesis and therapy. Annu. Rev. Med. 2007, 58, 157–170. [Google Scholar] [CrossRef]
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Invest. 2014, 124, 3047–3060. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Tang, X.X.; Ostedgaard, L.S.; Hoegger, M.J.; Moninger, T.O.; Karp, P.H.; McMenimen, J.D.; Choudhury, B.; Varki, A.; Stoltz, D.A.; Welsh, M.J. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J. Clin. Invest. 2016, 126, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Garcia, M.A.; Quinton, P.M. Normal mucus formation requires cAMP-dependent HCO3- secretion and Ca2+-mediated mucin exocytosis. J. Physiol. 2013, 591, 4581–4593. [Google Scholar] [CrossRef]
- Schultz, A.; Puvvadi, R.; Borisov, S.M.; Shaw, N.C.; Klimant, I.; Berry, L.J.; Montgomery, S.T.; Nguyen, T.; Kreda, S.M.; Kicic, A.; et al. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat. Commun. 2017, 8, 1409. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, S.H.; Bennett, W.D.; Zeman, K.L.; Knowles, M.R.; Tarran, R.; Boucher, R.C. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 2006, 354, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkins, M.R.; Robinson, M.; Rose, B.R.; Harbour, C.; Moriarty, C.P.; Marks, G.B.; Belousova, E.G.; Xuan, W.; Bye, P.T.; National Hypertonic Saline in Cystic Fibrosis (NHSCF) Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 2006, 354, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, M.T.; Flume, P.A. Cystic Fibrosis: Translating Molecular Mechanisms into Effective Therapies. Ann. Am. Thorac. Soc. 2018, 15, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Altes, T.A.; Johnson, M.; Fidler, M.; Botfield, M.; Tustison, N.J.; Leiva-Salinas, C.; de Lange, E.E.; Froh, D.; Mugler, J.P. 3rd. Use of hyperpolarized helium-3 MRI to assess response to ivacaftor treatment in patients with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.; Ehrhardt, A.; Hong, J.S.; Sorscher, E. Cystic fibrosis precision therapeutics: Emerging considerations. Pediatr. Pulmonol. 2019, 54, S13–S17. [Google Scholar] [CrossRef] [Green Version]
- Perrem, L.; Ratjen, F. Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr. Pulmonol. 2019, 54, S46–S55. [Google Scholar] [CrossRef] [Green Version]
- Regnis, J.A.; Robinson, M.; Bailey, D.L.; Cook, P.; Hooper, P.; Chan, H.K.; Gonda, I.; Bautovich, G.; Bye, P.T. Mucociliary clearance in patients with cystic fibrosis and in normal subjects. Am. J. Respir. Crit. Care Med. 1994, 150, 66–71. [Google Scholar] [CrossRef]
- Moore, P.J.; Tarran, R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert Opin. Ther. Targets 2018, 22, 687–701. [Google Scholar] [CrossRef]
- Kerem, E.; Bistritzer, T.; Hanukoglu, A.; Hofmann, T.; Zhou, Z.; Bennett, W.; MacLaughlin, E.; Barker, P.; Nash, M.; Quittell, L.; et al. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N. Engl. J. Med. 1999, 341, 156–162. [Google Scholar] [CrossRef]
- Köhler, D.; App, E.; Schmitz-Schumann, M.; Würtemberger, G.; Matthys, H. Inhalation of amiloride improves the mucociliary and the cough clearance in patients with cystic fibroses. Eur. J. Respir. Dis. Suppl. 1986, 146, 319–326. [Google Scholar]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef]
- Zhou, Z.; Duerr, J.; Johannesson, B.; Schubert, S.C.; Treis, D.; Harm, M.; Graeber, S.Y.; Dalpke, A.; Schultz, C.; Mall, M.A. The ENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J. Cyst. Fibros. 2011, 10, S172–S182. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, A.J. Altering airway surface liquid volume: Inhalation therapy with amiloride and hyperosmotic agents. Adv. Drug Deliv. Rev. 2002, 54, 1445–1462. [Google Scholar] [CrossRef]
- Perazella, M.A. Drug-induced hyperkalemia: Old culprits and new offenders. Am. J. Med. 2000, 109, 307–314. [Google Scholar] [CrossRef]
- Danahay, H.; McCarthy, C.; Gosling, M. A systematic comparison of the profiles of inhaled ENaC blocker candidates on mucociliary clearance: Are we under-dosing in clinical studies? J. Cyst. Fibros. 2019, 18, S41. [Google Scholar] [CrossRef]
- Avella, M.; Loriol, C.; Boulukos, K.; Borgese, F.; Ehrenfeld, J. SLC26A9 stimulates CFTR expression and function in human bronchial cell lines. J. Cell. Physiol. 2011, 226, 212–223. [Google Scholar] [CrossRef]
- Bertrand, C.A.; Zhang, R.; Pilewski, J.M.; Frizzell, R.A. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J. Gen. Physiol. 2009, 133, 421–438. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Salomon, J.J.; Sheppard, D.N.; Mall, M.A.; Galietta, L.J. Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr. Opin. Pharmacol. 2017, 34, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Anagnostopoulou, P.; Riederer, B.; Duerr, J.; Michel, S.; Binia, A.; Agrawal, R.; Liu, X.; Kalitzki, K.; Xiao, F.; Chen, M.; et al. SLC26A9-mediated chloride secretion prevents mucus obstruction in airway inflammation. J. Clin. Invest. 2012, 122, 3629–3634. [Google Scholar] [CrossRef] [Green Version]
- Gibson, A.; Lewis, A.P.; Affleck, K.; Aitken, A.J.; Meldrum, E.; Thompson, N. hCLCA1 and mCLCA3 are secreted non-integral membrane proteins and therefore are not ion channels. J. Biol. Chem. 2005, 280, 27205–27212. [Google Scholar] [CrossRef] [Green Version]
- Borsani, G.; Rugarli, E.; Taglialatela, M.; Wong, C.; Ballabio, A. Characterization of a human and murine gene (CLCN3) sharing similarities to voltage-gated chloride channels and to a yeast integral membrane protein. Genomics 1995, 27, 131–141. [Google Scholar] [CrossRef]
- Hartzell, H.C.; Qu, Z.; Yu, K.; Xiao, Q.; Chien, L.T. Molecular physiology of bestrophins: Multifunctional membrane proteins linked to best disease and other retinopathies. Physiol. Rev. 2008, 88, 639–672. [Google Scholar] [CrossRef]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.D.; Cho, H.; Koo, J.; Tak, M.H.; Cho, Y.; Shim, W.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Yurtsever, Z.; Berry, K.N.; Nichols, C.G.; Brett, T.J. Modulation of TMEM16A channel activity by the von Willebrand factor type A (VWA) domain of the calcium-activated chloride channel regulator 1 (CLCA1). J. Biol. Chem. 2017, 292, 9164–9174. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; O’Neal, W.K.; Gabriel, S.E.; Randell, S.H.; Harfe, B.D.; Boucher, R.C.; Grubb, B.R. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl− secretory channel in mouse airways. J. Biol. Chem. 2009, 284, 14875–14880. [Google Scholar] [CrossRef] [Green Version]
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A Potentiation: A Novel Therapeutic Approach for the Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Yerxa, B.R.; Sabater, J.R.; Davis, C.W.; Stutts, M.J.; Lang-Furr, M.; Picher, M.; Jones, A.C.; Cowlen, M.; Dougherty, R.; Boyer, J.; et al. Pharmacology of INS37217 [P(1)-(uridine 5′)-P(4)- (2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y(2) receptor agonist for the treatment of cystic fibrosis. J. Pharmacol. Exp. Ther. 2002, 302, 871–880. [Google Scholar] [CrossRef]
- Oliynyk, I.; Varelogianni, G.; Roomans, G.M.; Johannesson, M. Effect of duramycin on chloride transport and intracellular calcium concentration in cystic fibrosis and non-cystic fibrosis epithelia. APMIS 2010, 118, 982–990. [Google Scholar] [CrossRef]
- Billet, A.; Hanrahan, J.W. The secret life of CFTR as a calcium-activated chloride channel. J. Physiol. 2013, 591, 5273–5278. [Google Scholar] [CrossRef]
- Devor, D.C.; Pilewski, J.M. UTP inhibits Na+ absorption in wild-type and DeltaF508 CFTR-expressing human bronchial epithelia. Am. J. Physiol. 1999, 276, C827–CC837. [Google Scholar] [CrossRef]
- Lethem, M.I.; Dowell, M.L.; Van Scott, M.; Yankaskas, J.R.; Egan, T.; Boucher, R.C.; Davis, C.W. Nucleotide regulation of goblet cells in human airway epithelial explants: Normal exocytosis in cystic fibrosis. Am. J. Respir. Cell. Mol. Biol. 1993, 9, 315–322. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Dirksen, E.R. Mechanosensitive and beta-adrenergic control of the ciliary beat frequency of mammalian respiratory tract cells in culture. Am. Rev. Respir. Dis. 1989, 139, 432–440. [Google Scholar] [CrossRef]
- Moss, R.B. Pitfalls of drug development: Lessons learned from trials of denufosol in cystic fibrosis. J. Pediatr. 2013, 162, 676–680. [Google Scholar] [CrossRef]
- Grasemann, H.; Stehling, F.; Brunar, H.; Widmann, R.; Laliberte, T.W.; Molina, L.; Döring, G.; Ratjen, F. Inhalation of Moli1901 in patients with cystic fibrosis. Chest 2007, 131, 1461–1466. [Google Scholar] [CrossRef]
- Button, B.; Okada, S.F.; Frederick, C.B.; Thelin, W.R.; Boucher, R.C. Mechanosensitive ATP release maintains proper mucus hydration of airways. Sci. Signal. 2013, 6, ra46. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.L.; Harline, M.C.; Otero, M.A.; Glover, G.G.; Garrad, R.C.; Krugh, B.; Walker, N.M.; González, F.A.; Turner, J.T.; Weisman, G.A. Desensitization of P2Y2 receptor-activated transepithelial anion secretion. Am. J. Physiol. 1999, 276, C777–C787. [Google Scholar] [CrossRef]
- Luan, X.; Tam, J.S.; Belev, G.; Jagadeeshan, S.; Murray, B.; Hassan, N.; Machen, T.E.; Chapman, L.D.; Ianowski, J.P. Nebulized hypertonic saline triggers nervous system-mediated active liquid secretion in cystic fibrosis swine trachea. Sci. Rep. 2019, 9, 540. [Google Scholar] [CrossRef]
- Benedetto, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019, 33, 4502–4512. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X.; Wang, H.; Jiao, J.; Li, Y.; Fan, E.; Zhang, L.; Bachert, C. TMEM16A-Mediated Mucin Secretion in IL-13-Induced Nasal Epithelial Cells From Chronic Rhinosinusitis Patients. Allergy Asthma Immunol. Res. 2015, 7, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Jiang, Y.; Li, L.; Liu, Y.; Tang, H.; Jiang, D. TMEM16A mediates the hypersecretion of mucus induced by Interleukin-13. Exp. Cell. Res. 2015, 334, 260–269. [Google Scholar] [CrossRef]
- Qin, Y.; Jiang, Y.; Sheikh, A.S.; Shen, S.; Liu, J.; Jiang, D. Interleukin-13 stimulates MUC5AC expression via a STAT6-TMEM16A-ERK1/2 pathway in human airway epithelial cells. Int. Immunopharmacol. 2016, 40, 106–114. [Google Scholar] [CrossRef]
- Pedemonte, N.; Galietta, L.J. Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 2014, 94, 419–459. [Google Scholar] [CrossRef] [Green Version]
- Marty, A.; Tan, Y.P.; Trautmann, A. Three types of calcium-dependent channel in rat lacrimal glands. J. Physiol. 1984, 357, 293–325. [Google Scholar] [CrossRef] [Green Version]
- Willumsen, N.J.; Boucher, R.C. Activation of an apical Cl− conductance by Ca2+ ionophores in cystic fibrosis airway epithelia. Am. J. Physiol. 1989, 256, C226–C233. [Google Scholar] [CrossRef]
- Anderson, M.P.; Welsh, M.J. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc. Natl. Acad. Sci. USA 1991, 88, 6003–6007. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.J.; Paradiso, A.M.; Boucher, R.C. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. Br. J. Pharmacol. 1991, 103, 1649–1656. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Clarke, L.L.; Boucher, R.C. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N. Engl. J. Med. 1991, 325, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Tarran, R.; Button, B.; Picher, M.; Paradiso, A.M.; Ribeiro, C.M.; Lazarowski, E.R.; Zhang, L.; Collins, P.L.; Pickles, R.J.; Fredberg, J.J.; et al. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J. Biol. Chem. 2005, 280, 35751–35759. [Google Scholar] [CrossRef] [Green Version]
- Button, B.; Boucher, R.C. University of North Carolina Virtual Lung Group. Role of mechanical stress in regulating airway surface hydration and mucus clearance rates. Respir. Physiol. Neurobiol. 2008, 163, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Lazarowski, E.R.; Tarran, R.; Milgram, S.L.; Boucher, R.C.; Stutts, M.J. Compartmentalized autocrine signalling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14120–14125. [Google Scholar] [CrossRef] [Green Version]
- Lazarowski, E.R.; Tarran, R.; Grubb, B.R.; van Heusden, C.A.; Okada, S.; Boucher, R.C. Nucleotide release provides a mechanism for airway surface liquid homeostasis. J. Biol. Chem. 2004, 279, 36855–36864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, M.E.; Prasad, S.A. Physiotherapy management of cystic fibrosis. Chron. Respir. Dis. 2005, 2, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, C.M.; Baker, S.E.; Morgan, M.A.; Martinez, M.G.; Liu, B.; Rowe, S.M.; Morgan, W.J.; Wong, E.C.; Karpen, S.R.; Snyder, E.M. Moderate intensity exercise mediates comparable increases in exhaled chloride as albuterol in individuals with cystic fibrosis. Respir. Med. 2015, 109, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Clarke, L.L.; Boucher, R.C. Extracellular ATP and UTP induce chloride secretion in nasal epithelia of cystic fibrosis patients and normal subjects in vivo. Chest 1992, 101, 60S–63S. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.L.; Boucher, R.C. Chloride secretory response to extracellular ATP in human normal and cystic fibrosis nasal epithelia. Am. J. Physiol. 1992, 263, C348–C356. [Google Scholar] [CrossRef]
- Paradiso, A.M.; Ribeiro, C.M.; Boucher, R.C. Polarized signalling via purinoceptors in normal and cystic fibrosis airway epithelia. J. Gen. Physiol. 2001, 117, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.M.; Paradiso, A.M.; Carew, M.A.; Shears, S.B.; Boucher, R.C. Cystic fibrosis airway epithelial Ca2+ i signalling: The mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 10202–10209. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Sun, L.; Kato, T.; Okuda, K.; Martino, M.B.; Abzhanova, A.; Lin, J.M.; Gilmore, R.C.; Batson, B.D.; O’Neal, Y.K.; et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J. Clin. Invest. 2019, 129, 4433–4450. [Google Scholar] [CrossRef] [PubMed]
- Balghi, H.; Robert, R.; Rappaz, B.; Zhang, X.; Wohlhuter-Haddad, A.; Evagelidis, A.; Luo, Y.; Goepp, J.; Ferraro, P.; Roméo, P.; et al. Enhanced Ca2+ entry due to Orai1 plasma membrane insertion increases IL-8 secretion by cystic fibrosis airways. FASEB J. 2011, 25, 4274–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atherton, H.; Mesher, J.; Poll, C.T.; Danahay, H. Preliminary pharmacological characterisation of an interleukin-13-enhanced calcium-activated chloride conductance in the human airway epithelium. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 367, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Caci, E.; Scudieri, P.; Di Carlo, E.; Morelli, P.; Bruno, S.; De Fino, I.; Bragonzi, A.; Gianotti, A.; Sondo, E.; Ferrera, L.; et al. Upregulation of TMEM16A Protein in Bronchial Epithelial Cells by Bacterial Pyocyanin. PLoS ONE 2015, 10, e0131775. [Google Scholar] [CrossRef]
- Scudieri, P.; Caci, E.; Bruno, S.; Ferrera, L.; Schiavon, M.; Sondo, E.; Tomati, V.; Gianotti, A.; Zegarra-Moran, O.; Pedemonte, N.; et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J. Physiol. 2012, 590, 6141–6155. [Google Scholar] [CrossRef] [PubMed]
- Atherton, H.C.; Jones, G.; Danahay, H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L730–L739. [Google Scholar] [CrossRef] [Green Version]
- Dolganov, G.M.; Woodruff, P.G.; Novikov, A.A.; Zhang, Y.; Ferrando, R.E.; Szubin, R.; Fahy, J.V. A novel method of gene transcript profiling in airway biopsy homogenates reveals increased expression of a Na+-K+-Cl− cotransporter (NKCC1) in asthmatic subjects. Genome Res. 2001, 11, 1473–1483. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, I.; Benedetto, R.; Fonseca, A.; Wanitchakool, P.; Sirianant, L.; Skryabin, B.V.; Schenk, L.K.; Pavenstädt, H.; Schreiber, R.; Kunzelmann, K. Differential effects of anoctamins on intracellular calcium signals. FASEB J. 2017, 31, 2123–2134. [Google Scholar] [CrossRef]
- Schreiber, R.; Ousingsawat, J.; Wanitchakool, P.; Sirianant, L.; Benedetto, R.; Reiss, K.; Kunzelmann, K. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid. J. Physiol. 2018, 596, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Sabater, J.R.; Mao, Y.M.; Shaffer, C.; James, M.K.; O’Riordan, T.G.; Abraham, W.M. Aerosolization of P2Y(2)-receptor agonists enhances mucociliary clearance in sheep. J. Appl. Physiol. 1999, 87, 2191–2196. [Google Scholar] [CrossRef] [Green Version]
- Bennett, W.D.; Zeman, K.L.; Foy, C.; Shaffer, C.L.; Johnson, F.L.; Regnis, J.A.; Sannuti, A.; Johnson, J. Effect of aerosolized uridine 5′-triphosphate on mucociliary clearance in mild chronic bronchitis. Am. J. Respir. Crit. Care Med. 2001, 164, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Ousingsawat, J.; Cabrita, I.; Doušová, T.; Bähr, A.; Janda, M.; Schreiber, R.; Benedetto, R. TMEM16A in Cystic Fibrosis: Activating or Inhibiting? Front. Pharmacol. 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Rock, J.R.; Harfe, B.D.; Cheng, T.; Huang, X.; Jan, Y.N.; Jan, L.Y. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc. Natl. Acad. Sci. USA 2009, 106, 21413–21418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallos, G.; Remy, K.E.; Danielsson, J.; Funayama, H.; Fu, X.W.; Chang, H.Y.; Yim, P.; Xu, D.; Emala, C.W., Sr. Functional expression of the TMEM16 family of calcium-activated chloride channels in airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L625–L634. [Google Scholar] [CrossRef] [Green Version]
- Miner, K.; Labitzke, K.; Liu, B.; Wang, P.; Henckels, K.; Gaida, K.; Elliott, R.; Chen, J.J.; Liu, L.; Leith, A.; et al. Drug Repurposing: The Anthelmintics Niclosamide and Nitazoxanide Are Potent TMEM16A Antagonists That Fully Bronchodilate Airways. Front. Pharmacol. 2019, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Amaral, M.D.; Beekman, J.M. Activating alternative chloride channels to treat CF: Friends or Foes?: Report on the Meeting of the Basic Science Working Group in Dubrovnik, Croatia. J. Cyst. Fibros. 2020, 19, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Simões, F.B.; Quaresma, M.C.; Clarke, L.A.; Silva, I.A.; Pankonien, I.; Railean, V.; Kmit, A.; Amaral, M.D. TMEM16A chloride channel does not drive mucus production. Life Sci. Alliance 2019, 15, e201900462. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Wang, Y.; Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Correctors promote maturation of cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by binding to the protein. J. Biol. Chem. 2007, 282, 33247–33251. [Google Scholar] [CrossRef] [Green Version]
- Felser, A.; Lindinger, P.W.; Schnell, D.; Kratschmar, D.V.; Odermatt, A.; Mies, S.; Jenö, P.; Krähenbühl, S. Hepatocellular toxicity of benzbromarone: Effects on mitochondrial function and structure. Toxicology 2014, 324, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G.I.; Jin, S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med. 2014, 20, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott-Ward, T.S.; Li, H.; Schmidt, A.; Cai, Z.; Sheppard, D.N. Direct block of the cystic fibrosis transmembrane conductance regulator Cl(-) channel by niflumic acid. Mol. Membr. Biol. 2004, 21, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.E.; Herzer, T.; Wagner, C.A.; Schmidt, F.; Raber, G.; Waldegger, S.; Lang, F. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol. Pharmacol. 1994, 46, 750–753. [Google Scholar]
- Hu, H.; Tian, J.; Zhu, Y.; Wang, C.; Xiao, R.; Herz, J.M.; Wood, J.D.; Zhu, M.X. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Arch. 2010, 459, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.L.; Koh, S.D.; Sergeant, G.P.; Sanders, K.M.; Horowitz, B. TRPC4 currents have properties similar to the pacemaker current in interstitial cells of Cajal. Am. J. Physiol. Cell Physiol. 2002, 283, C1637–C1645. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Lee, H.K.; Park, J.; Jeon, D.K.; Jo, S.; Jo, M.; Namkung, W. Ani9, A Novel Potent Small-Molecule ANO1 Inhibitor with Negligible Effect on ANO2. PLoS ONE 2016, 11, e0155771. [Google Scholar] [CrossRef] [Green Version]
- Bill, A.; Gaither, L. The Mechanistic Role of the Calcium-Activated Chloride Channel ANO1 in Tumor Growth and Signalling. Adv. Exp. Med. Biol. 2017, 966, 1–14. [Google Scholar]
- Leblanc, N.; Forrest, A.S.; Ayon, R.J.; Wiwchar, M.; Angermann, J.E.; Pritchard, H.A.; Singer, C.A.; Valencik, M.L.; Britton, F.; Greenwood, I.A. Molecular and functional significance of Ca(2+)-activated Cl(-) channels in pulmonary arterial smooth muscle. Pulm. Circ. 2015, 5, 244–268. [Google Scholar] [CrossRef] [Green Version]
- Heinze, C.; Seniuk, A.; Sokolov, M.V.; Huebner, A.K.; Klementowicz, A.E.; Szijártó, I.A.; Schleifenbaum, J.; Vitzthum, H.; Gollasch, M.; Ehmke, H.; et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J. Clin. Invest. 2014, 124, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, D.M.; Kim, S.; Jensen, A.B.; Matchkov, V.M.; Andersson, K.E. New selective inhibitors of calcium-activated chloride channels - T16A(inh) -A01, CaCC(inh) -A01 and MONNA - what do they inhibit? Br. J. Pharmacol. 2015, 172, 4158–4172. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, C.; Huai, R.; Qu, Z. Overexpression of ANO1/TMEM16A, an arterial Ca2+-activated Cl− channel, contributes to spontaneous hypertension. J. Mol. Cell. Cardiol. 2015, 82, 22–32. [Google Scholar] [CrossRef]
- Forrest, A.S.; Joyce, T.C.; Huebner, M.L.; Ayon, R.J.; Wiwchar, M.; Joyce, J.; Freitas, N.; Davis, A.J.; Ye, L.; Duan, D.D.; et al. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2012, 303, C1229–C1243. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.L.; Sun, L.; Zheng, H.Q.; Wang, G.L.; Du, Y.H.; Lv, X.F.; Ma, M.M.; Guan, Y.Y. Smooth muscle-specific TMEM16A expression protects against angiotensin II-induced cerebrovascular remodeling via suppressing extracellular matrix deposition. J. Mol. Cell. Cardiol. 2019, 134, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Cil, O.; Anderson, M.; Yen, R.; Kelleher, B.; Huynh, T.L.; Seo, Y.; Nilsen, S.P.; Turner, J.R.; Verkman, A.S. Slowed gastric emptying and improved oral glucose tolerance produced by a nanomolar-potency inhibitor of calcium-activated chloride channel TMEM16A. FASEB J. 2019, 33, 11247–11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ousingsawat, J.; Martins, J.R.; Schreiber, R.; Rock, J.R.; Harfe, B.D.; Kunzelmann, K. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J. Biol. Chem. 2009, 284, 28698–28703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Yang, Y.D.; Lee, J.; Lee, B.; Kim, T.; Jang, Y.; Back, S.K.; Na, H.S.; Harfe, B.D.; Wang, F.; et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat. Neurosci. 2012, 15, 1015–1021. [Google Scholar] [CrossRef]
- Lee, B.; Cho, H.; Jung, J.; Yang, Y.D.; Yang, D.J.; Oh, U. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol. Pain 2014, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.Y.; Tan, C.Y.; Wang, Y.; Ma, K.T.; Li, L.; Si, J.Q. Mechanism of persistent hyperalgesia in neuropathic pain caused by chronic constriction injury. Neural Regen. Res. 2019, 14, 1091–1098. [Google Scholar]
- Seo, K.H.; Jin, Y.; Jung, S.Y.; Lee, S.H. Comprehensive behavioral analyses of anoctamin1/TMEM16A-conditional knockout mice. Life Sci. 2018, 207, 323–331. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Jiang, X.; Chan, H. Cystic fibrosis transmembrane conductance regulator-emerging regulator of cancer. Cell. Mol. Life Sci. 2018, 75, 1737–1756. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Guo, S.; Wang, X.; Pang, C.; Zhan, Y.; Chen, Y.; An, H. Recent advances in TMEM16A: Structure, function, and disease. J. Cell. Physiol. 2019, 234, 7856–7873. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Song, Y.; Gao, J.; Gao, J.; Wang, K. Inhibition of calcium-activated chloride channel ANO1 suppresses proliferation and induces apoptosis of epithelium originated cancer cells. Oncotarget 2016, 7, 78619–78630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill, A.; Hall, M.L.; Borawski, J.; Hodgson, C.; Jenkins, J.; Piechon, P.; Popa, O.; Rothwell, C.; Tranter, P.; Tria, S.; et al. Small molecule-facilitated degradation of ANO1 protein: A new targeting approach for anticancer therapeutics. J. Biol. Chem. 2014, 289, 11029–11041. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danahay, H.; Gosling, M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? Int. J. Mol. Sci. 2020, 21, 2386. https://doi.org/10.3390/ijms21072386
Danahay H, Gosling M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? International Journal of Molecular Sciences. 2020; 21(7):2386. https://doi.org/10.3390/ijms21072386
Chicago/Turabian StyleDanahay, Henry, and Martin Gosling. 2020. "TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis?" International Journal of Molecular Sciences 21, no. 7: 2386. https://doi.org/10.3390/ijms21072386
APA StyleDanahay, H., & Gosling, M. (2020). TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? International Journal of Molecular Sciences, 21(7), 2386. https://doi.org/10.3390/ijms21072386