Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi®) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells


 , ,
, , 
Abstract
:1. Introduction
2. Results
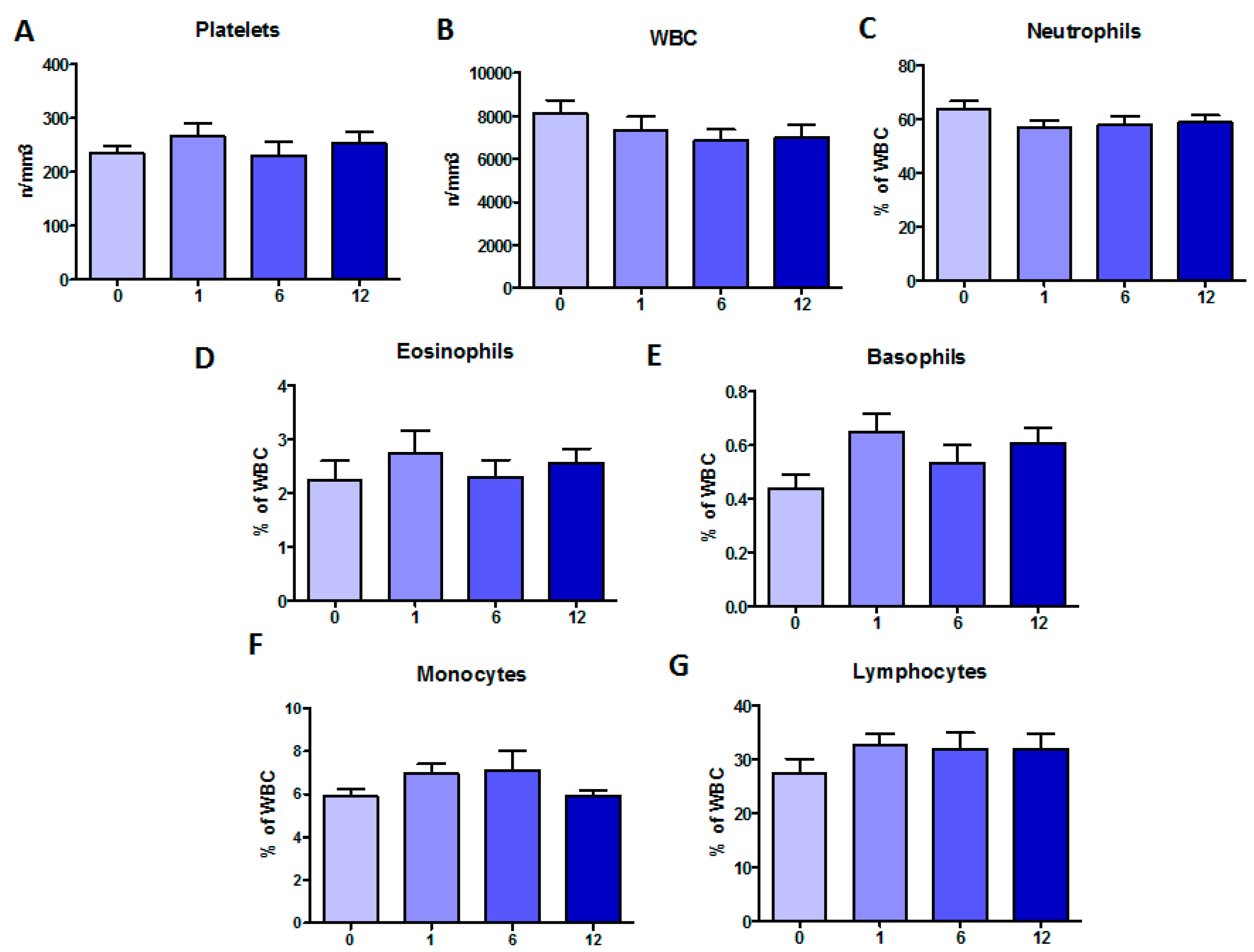
2.1. Clinical Monitoring during Orkambi® Treatment
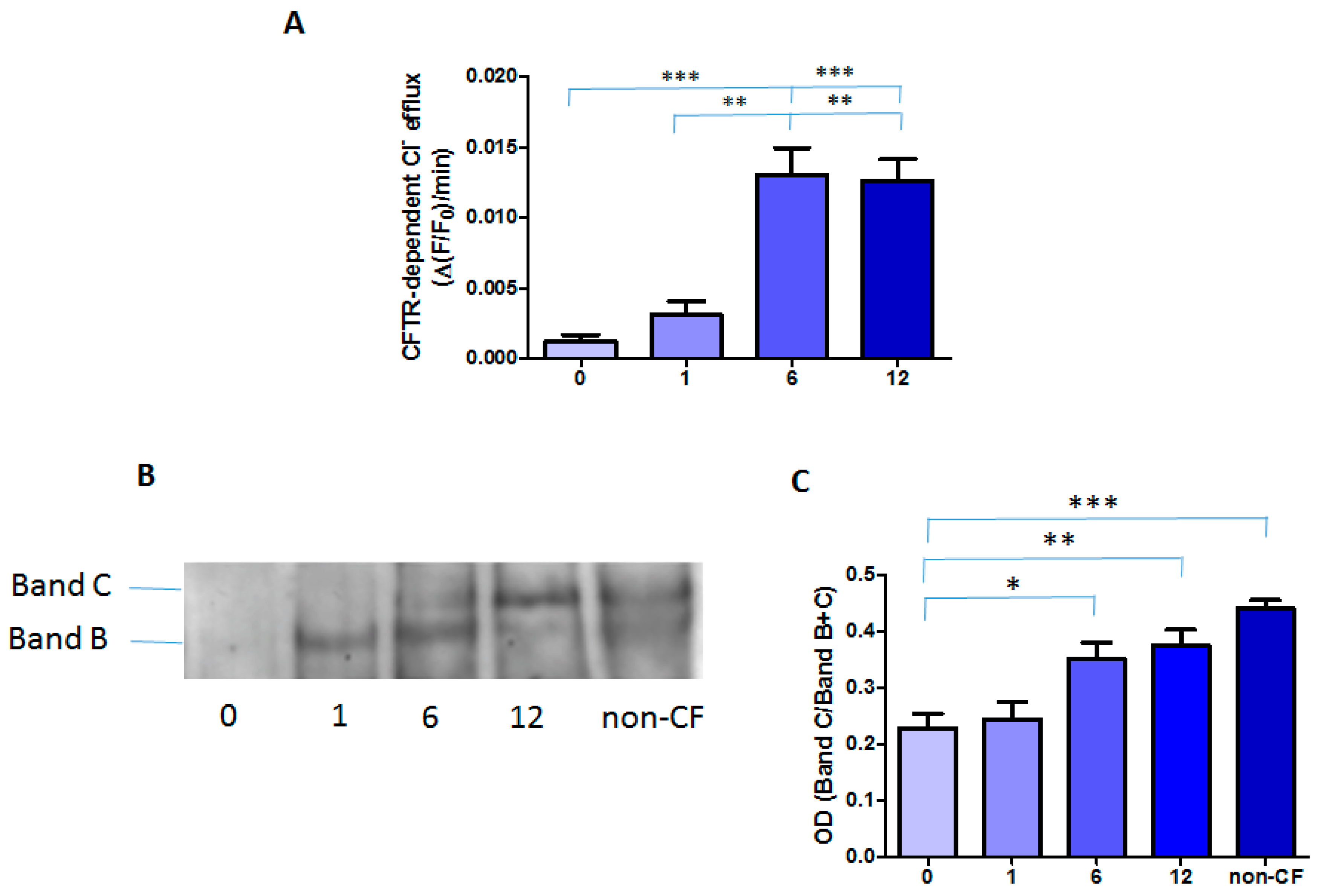
2.2. Effect of the Treatment with Orkambi® on CFTR-Dependent Chloride Efflux and Protein in MNCs
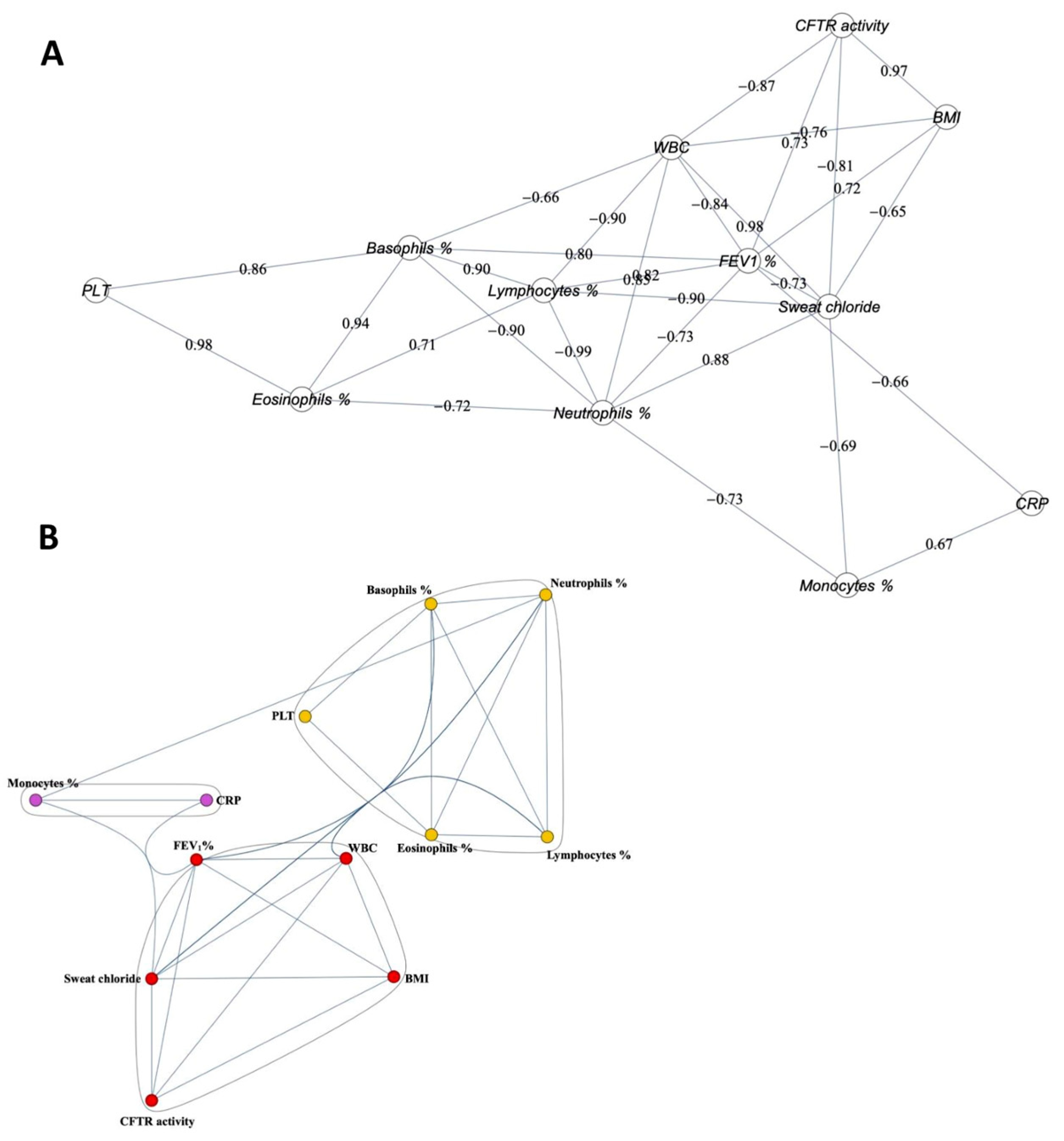
2.3. Correlation among the Clinical Parameters and CFTR Activity
3. Discussion
4. Patients and Methods
4.1. Patients
4.2. Isolation of Blood Mononuclear Cells
4.3. CFTR Function Measurement in Mononuclear Cells
4.4. Western Blot of CFTR in MNCs
4.5. Statistical analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| BMI | Body mass index |
| CRP | C-reactive protein |
| FEV1 | Forced expiratory volume in 1 s |
References
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Gentzsch, M.; Chang, X.B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Lukacs, G.L.; Segal, G.; Kartner, N.; Grinstein, S.; Zhang, F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem. J. 1997, 328 Pt 2, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Denning, G.M.; Ostedgaard, L.S.; Welsh, M.J. Abnormal localization of cystic fibrosis transmembrane conductance regulator in prymary cultures of cystic fibrosis airway epithelia. J. Cell Biol. 1992, 118, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Sharma, M.; Lukacs, G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- Turnbull, E.L.; Rosser, M.F.; Cyr, D.M. The role of the UPS in cystic fibrosis. BMC Biochem. 2007, 8 (Suppl. S1), S11. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef]
- Boucher, R.C. Cystic fibrosis: A disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007, 13, 231–240. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckford, P.D.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K.; et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 2014, 21, 666–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.; Houwen, R.H.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra384. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, N.; Moreira, T.; Michel, P.; Steiner, T.; Jansen, O.; Cognard, C.; Mattle, H.P.; van Zwam, W.; Holmin, S.; Tatlisumak, T.; et al. Mechanical thrombectomy in acute ischemic stroke: Consensus statement by ESO-Karolinska Stroke Update 2014/2015, supported by ESO, ESMINT, ESNR and EAN. Int. J. Stroke 2016, 11, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Milla, C.E.; Ratjen, F.; Marigowda, G.; Liu, F.; Waltz, D.; Rosenfeld, M.; VX13-809-011 Part B Investigator Group. Lumacaftor/ivacaftor in patients aged 6–11 years with cystic fibrosis and homozygous for F508del-CFTR. Am. J. Respir. Crit. Care Med. 2017, 195, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra296. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Wang, Y.; Wang, X.; Wrennall, J.A.; Rimington, T.L.; Li, H.; Cai, Z.; Ford, R.C.; Sheppard, D.N. Two small molecules restore stability to a subpopulation of the cystic fibrosis transmembrane conductance regulator with the predominant disease-causing mutation. J. Biol. Chem. 2017, 292, 3706–3719. [Google Scholar] [CrossRef] [Green Version]
- Masson, A.; Schneider-Futschik, E.K.; Baatallah, N.; Nguyen-Khoa, T.; Girodon, E.; Hatton, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; et al. Predictive factors for lumacaftor/ivacaftor clinical response. J. Cyst. Fibros. 2019, 18, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; D’Oria, S.; Favia, M.; Castellani, S.; Santostasi, T.; Polizzi, A.M.; Mariggio, M.A.; Gallo, C.; Casavola, V.; Montemurro, P.; et al. CFTR-dependent chloride efflux in cystic fibrosis mononuclear cells is increased by ivacaftor therapy. Pediatr. Pulmonol. 2017, 52, 900–908. [Google Scholar] [CrossRef]
- Guerra, L.; Favia, M.; Castellani, S.; Barbuti, G.; Montemurro, P.; Diana, A.; Santostasi, T.; Polizzi, A.M.; Mariggio, M.A.; Reshkin, S.J.; et al. Antibiotic therapy affects functional behaviour in cystic fibrosis blood mononuclear cells. Eur. Respir. J. 2015, 46, 558–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeev, V.; Chou, F.Y.; Lam, G.Y.; Hamilton, C.M.; Wilcox, P.G.; Quon, B.S. The extrapulmonary effects of cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Ann. Am. Thorac. Soc. 2020, 17, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Zhang, S.; Shrestha, C.L.; Kopp, B.T. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 17066. [Google Scholar] [CrossRef] [Green Version]
- White, M.M.; Geraghty, P.; Hayes, E.; Cox, S.; Leitch, W.; Alfawaz, B.; Lavelle, G.M.; McElvaney, O.J.; Flannery, R.; Keenan, J.; et al. Neutrophil membrane cholesterol content is a key factor in cystic fibrosis lung disease. EBioMedicine 2017, 23, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Hisert, K.B.; Schoenfelt, K.Q.; Cooke, G.; Grogan, B.; Launspach, J.L.; Gallagher, C.G.; Donnelly, S.C.; Welsh, M.J.; Singh, P.K.; McKone, E.F.; et al. Ivacaftor-induced proteomic changes suggest monocyte defects may contribute to the pathogenesis of cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 54, 594–597. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Kerem, E.; Reisman, J.; Corey, M.; Canny, G.J.; Levison, H. Prediction of mortality in patients with cystic fibrosis. N. Engl. J. Med. 1992, 326, 1187–1191. [Google Scholar] [CrossRef]
- Jennings, M.T.; Dezube, R.; Paranjape, S.; West, N.E.; Hong, G.; Braun, A.; Grant, J.; Merlo, C.A.; Lechtzin, N. An observational study of outcomes and tolerances in patients with cystic fibrosis initiated on lumacaftor/ivacaftor. Ann. Am. Thorac. Soc. 2017, 14, 162–1666. [Google Scholar] [CrossRef] [PubMed]
- Hubert, D.; Chiron, R.; Camara, B.; Grenet, D.; Prevotat, A.; Bassinet, L.; Dominique, S.; Rault, G.; Macey, J.; Honore, I.; et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J. Cyst. Fibros. 2017, 16, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Del Porto, P.; Cifani, N.; Guarnieri, S.; Di Domenico, E.G.; Mariggio, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar]
- Van de Weert-van Leeuwen, P.B.; Van Meegen, M.A.; Speirs, J.J.; Pals, D.J.; Rooijakkers, S.H.; Van der Ent, C.K.; Terheggen-Lagro, S.W.; Arets, H.G.; Beekman, J.M. Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am. J. Respir. Cell Mol. Biol. 2013, 49, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Hodges, C.A.; Cotton, C.U.; Drumm, M.L. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J. Leukoc. Biol. 2012, 92, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Min. Value | Median | Max. Value | 25th Percentile | 75th Percentile | |
|---|---|---|---|---|---|
| Sweat chloride (mEq/L) | 97.0 | 123.5 | 139.0 | 116.5 | 132.0 |
| FEV1% | 21.5 | 51.4 | 101.6 | 39.0 | 88.3 |
| BMI (kg/m2) | 16.7 | 20.6 | 23.9 | 18.5 | 21.8 |
| CRP (mg/dL) | 2.9 | 3.9 | 18.5 | 2.9 | 13.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favia, M.; Gallo, C.; Guerra, L.; De Venuto, D.; Diana, A.; Polizzi, A.M.; Montemurro, P.; Mariggiò, M.A.; Leonetti, G.; Manca, A.; et al. Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi®) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells. Int. J. Mol. Sci. 2020, 21, 2398. https://doi.org/10.3390/ijms21072398
Favia M, Gallo C, Guerra L, De Venuto D, Diana A, Polizzi AM, Montemurro P, Mariggiò MA, Leonetti G, Manca A, et al. Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi®) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells. International Journal of Molecular Sciences. 2020; 21(7):2398. https://doi.org/10.3390/ijms21072398
Chicago/Turabian StyleFavia, Maria, Crescenzio Gallo, Lorenzo Guerra, Domenica De Venuto, Anna Diana, Angela Maria Polizzi, Pasqualina Montemurro, Maria Addolorata Mariggiò, Giuseppina Leonetti, Antonio Manca, and et al. 2020. "Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi®) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells" International Journal of Molecular Sciences 21, no. 7: 2398. https://doi.org/10.3390/ijms21072398
APA StyleFavia, M., Gallo, C., Guerra, L., De Venuto, D., Diana, A., Polizzi, A. M., Montemurro, P., Mariggiò, M. A., Leonetti, G., Manca, A., Casavola, V., & Conese, M. (2020). Treatment of Cystic Fibrosis Patients Homozygous for F508del with Lumacaftor-Ivacaftor (Orkambi®) Restores Defective CFTR Channel Function in Circulating Mononuclear Cells. International Journal of Molecular Sciences, 21(7), 2398. https://doi.org/10.3390/ijms21072398