Tumor Milieu Controlled by RB Tumor Suppressor

Abstract
:
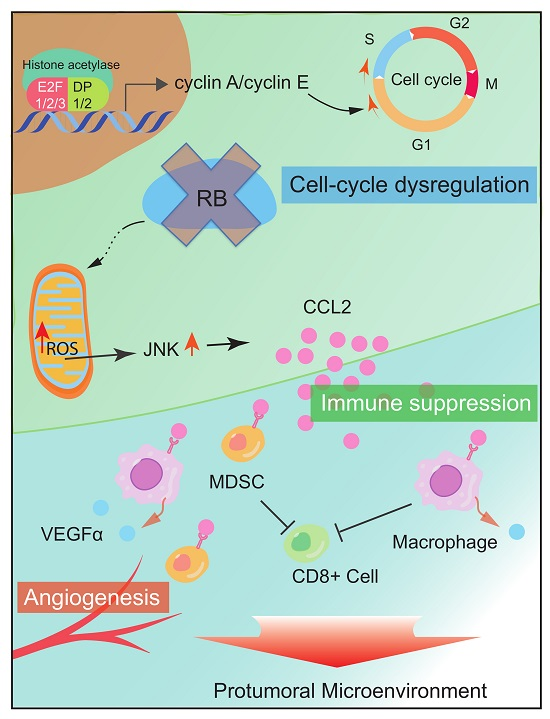{kind=link}
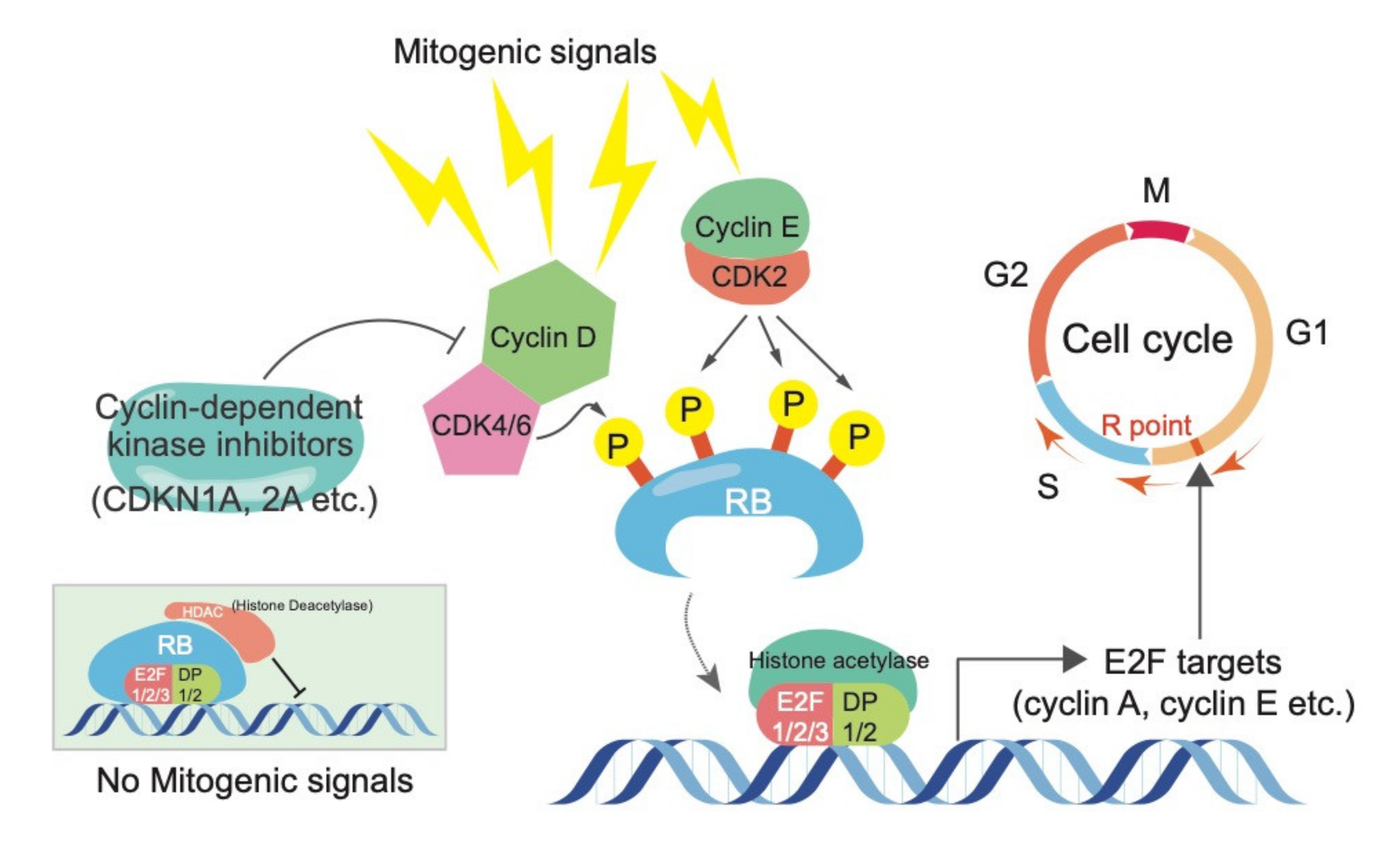{kind=link}
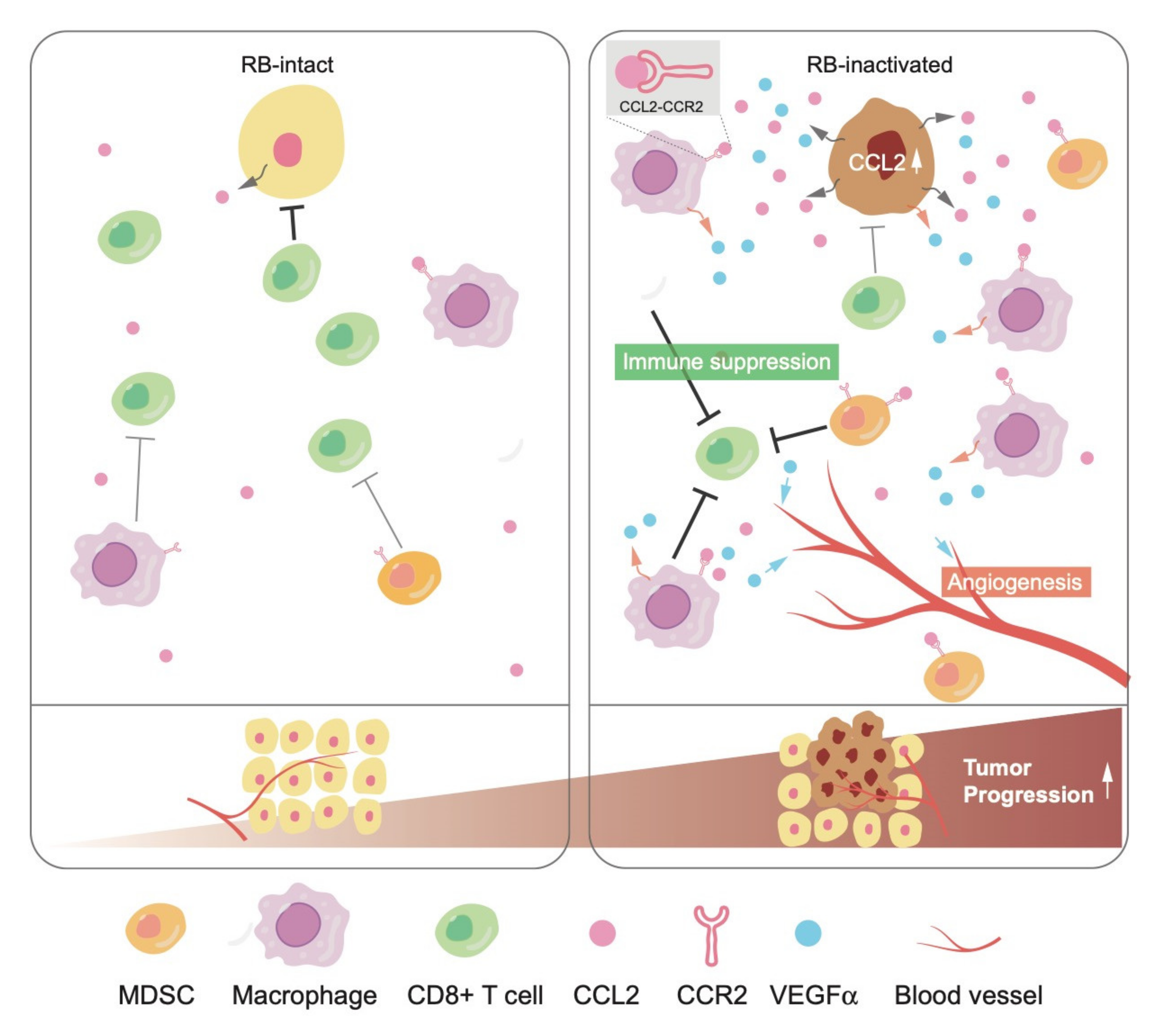{kind=link}
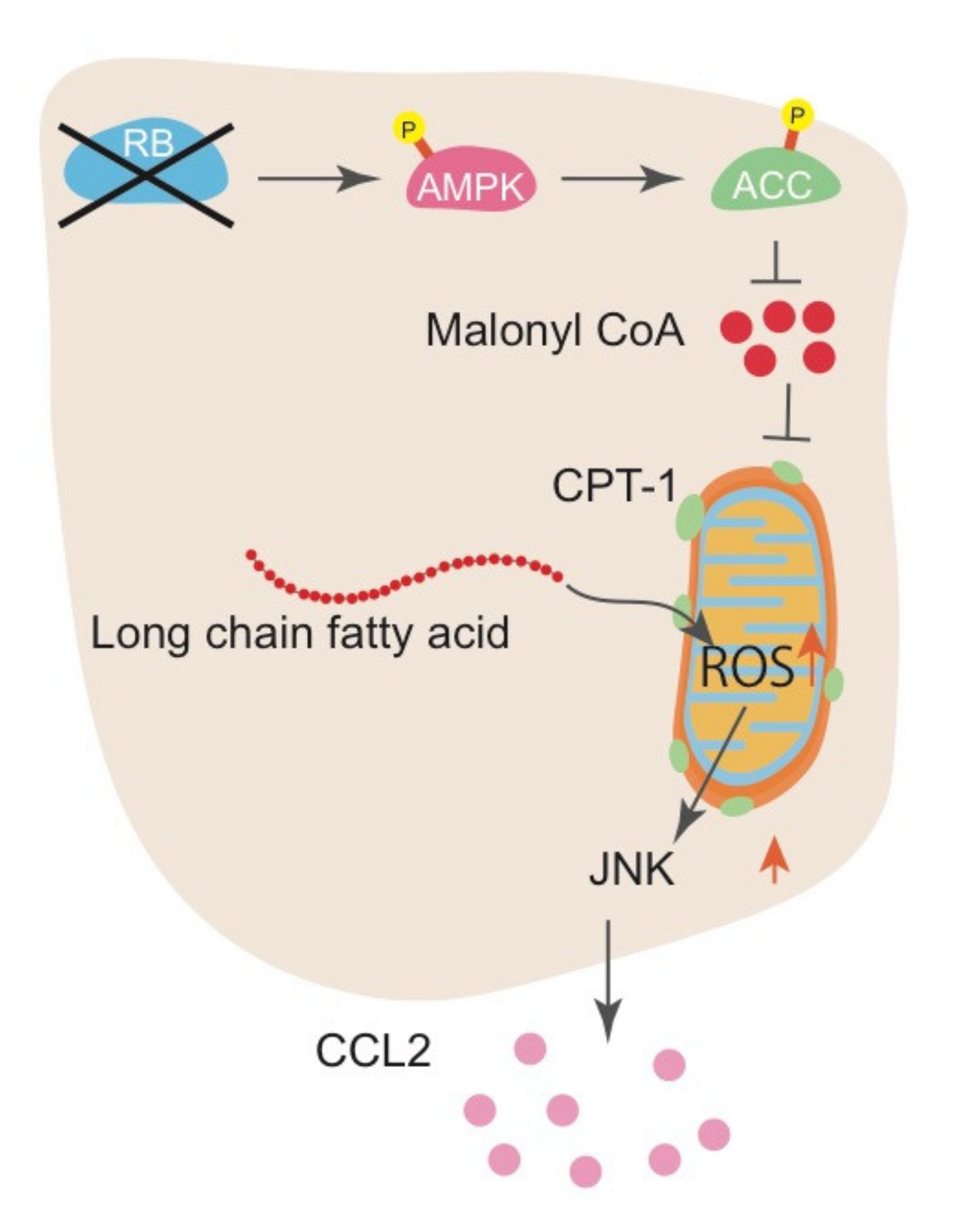{kind=link}
1. Introduction
2. Beyond Cell Cycle Regulation
2.1. Increased Lineage Plasticity Induced by RB Inactivation
2.2. Metabolic Reprogramming Induced by RB Inactivation
3. Enhanced Cytokines Secretion by Oxidative Stress Induced by RB
3.1. Increased Cellular Stress Induced by RB Inactivation Promotes Cytokine and Chemokine Secretion
3.2. RB Is Involved in the Mechanism to Keep Redox Balance at a Nontoxic Level
4. RB Impacts the Tumor Microenvironment via Chemokine
5. Therapeutic Strategy Targeting Innate Immune Signaling in RB-Inactive Cancer
6. Role of Other Tumor Suppressor Proteins in the TME
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDK | Cyclin-dependent kinase |
| HER | Human EGFR-related |
| TCA | Tricarboxylic acid |
| KDM | Lysine (K) Demethylase |
| ATP | Adenosine triphosphate |
| AMP | Adenosine monophosphate |
| IL- | Interleukin- |
| CCL | C-C motif ligand |
| CCR | C-C chemokine receptor |
| CXCL | C-X-C motif ligand |
| CoA | Coenzyme A |
| UTR | Untranslated region |
| MMTV | Mouse mammary tumor virus |
| JNK | c-jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| IFN | Interferon |
References
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Knudsen, K.E. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat. Rev. Cancer 2008, 8, 714–724. [Google Scholar] [CrossRef]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, L.; Lukas, J.; Bartkova, J.; Kjerulff, A.A.; Strauss, M.; Bartek, J. Aberrations of p16Ink4 and retinoblastoma tumour-suppressor genes occur in distinct sub-sets of human cancer cell lines. Int. J. Cancer 1995, 61, 115–120. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle. 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Johnson, J.; Thijssen, B.; McDermott, U.; Garnett, M.; Wessels, L.F.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host. Microbe. 2014, 16, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Coschi, C.H.; Ishak, C.A.; Gallo, D.; Marshall, A.; Talluri, S.; Wang, J.; Cecchini, M.J.; Martens, A.L.; Percy, V.; Welch, I.; et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov. 2014, 4, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [Green Version]
- Sage, J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 2012, 26, 1409–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, B.N.; Dyson, N.J. The multiple connections between pRB and cell metabolism. Curr. Opin. Cell. Biol. 2013, 25, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benevolenskaya, E.V.; Frolov, M.V. Emerging Links between E2F Control and Mitochondrial Function. Cancer Res. 2015, 75, 619–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.D.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.; Witkiewicz, A.K.; Knudsen, E.S. The RB tumor suppressor at the intersection of proliferation and immunity: Relevance to disease immune evasion and immunotherapy. Cell. Cycle 2015, 14, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Deng, T.; Jones, R.; Li, H.; Herschkowitz, J.I.; Liu, J.C.; Weigman, V.J.; Tsao, M.S.; Lane, T.F.; Perou, C.M.; et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Investig. 2010, 120, 3296–3309. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.M.; Yates, T.J.; Ruiz-Torres, M.; Kim-Kiselak, C.; Gudiel, A.A.; Deshpande, C.; Wang, W.Z.; Cicchini, M.; Stokes, K.L.; Tobias, J.W.; et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 2019, 569, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Inoue, Y.; Shibata, T.; Hayashi, H.; Nagano, O.; Saya, H.; Taya, Y. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res. 2008, 68, 5104–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, E.E.; Wang, Y.; Xu, H.; Zilfou, J.T.; Knudsen, K.E.; Aronow, B.J.; Lowe, S.W.; Knudsen, E.S. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J. Clin. Investig. 2007, 117, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Clem, B.; Zuba-Surma, E.K.; El-Naggar, S.; Telang, S.; Jenson, A.B.; Wang, Y.; Shao, H.; Ratajczak, M.Z.; Chesney, J.; et al. Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 2009, 4, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Pajcini, K.V.; Corbel, S.Y.; Sage, J.; Pomerantz, J.H.; Blau, H.M. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 2010, 7, 198–213. [Google Scholar]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Kohno, S.; Kondoh, A.; Sasaki, N.; Nishimoto, Y.; Li, F.; Abdallah Mohammed, M.S.; Muranaka, H.; Nagatani, N.; Suzuki, M.; et al. Undifferentiated State Induced by Rb-p53 Double Inactivation in Mouse Thyroid Neuroendocrine Cells and Embryonic Fibroblasts. Stem Cells 2015, 33, 1657–1669. [Google Scholar]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef]
- Shamma, A.; Takegami, Y.; Miki, T.; Kitajima, S.; Noda, M.; Obara, T.; Okamoto, T.; Takahashi, C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 2009, 15, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, M.; Vogel, H.; Lee, E.Y.; Bradley, A.; Donehower, L.A. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res. 1995, 55, 1146–1151. [Google Scholar] [PubMed]
- Williams, B.O.; Remington, L.; Albert, D.M.; Mukai, S.; Bronson, R.T.; Jacks, T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 1994, 7, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell. Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, B.N.; Gameiro, P.A.; Tschop, K.; Korenjak, M.; Heilmann, A.M.; Asara, J.M.; Stephanopoulos, G.; Iliopoulos, O.; Dyson, N.J. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013, 27, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.Y.; Hung, C.L.; Chen, Y.R.; Yang, J.C.; Wang, J.; Campbell, M.; Izumiya, Y.; Chen, H.W.; Wang, W.C.; Ann, D.K.; et al. KDM4A Coactivates E2F1 to Regulate the PDK-Dependent Metabolic Switch between Mitochondrial Oxidation and Glycolysis. Cell Rep. 2016, 16, 3016–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; De Milito, A.; Demiroglu-Zergeroglu, A.; Gullbo, J.; D’Arcy, P.; Linder, S. Eradicating Quiescent Tumor Cells by Targeting Mitochondrial Bioenergetics. Trends Cancer 2016, 2, 657–663. [Google Scholar] [CrossRef]
- Jones, R.A.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Investig. 2016, 126, 3739–3757. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Martin, V.; Gomez-Manzano, C.; Johnson, D.G.; Alonso, M.; White, E.; Xu, J.; McDonnell, T.J.; Shinojima, N.; Fueyo, J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010, 70, 7882–7893. [Google Scholar] [CrossRef] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [Green Version]
- Ciavarra, G.; Zacksenhaus, E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J. Cell Biol. 2010, 191, 291–301. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, B.N.; Danielian, P.S.; Kottakis, F.; Lapek, J.D., Jr.; Sanidas, I.; Miles, W.O.; Dehnad, M.; Tschop, K.; Gierut, J.J.; Manning, A.L.; et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. 2015, 29, 1875–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, M.L.; Berman, H.K.; Miller, C.; Kozakeiwicz, K.; Chew, K.; Moore, D.; Rabban, J.; Chen, Y.Y.; Kerlikowske, K.; Tlsty, T.D. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell 2007, 12, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiz-Ladera, C.; Lara, M.F.; Garin, M.; Ruiz, S.; Santos, M.; Lorz, C.; Garcia-Escudero, R.; Martinez-Fernandez, M.; Bravo, A.; Fernandez-Capetillo, O.; et al. p21 suppresses inflammation and tumorigenesis on pRB-deficient stratified epithelia. Oncogene 2014, 33, 4599–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Hinohara, K.; Kobayashi, S.; Kanauchi, H.; Shimizu, S.; Nishioka, K.; Tsuji, E.; Tada, K.; Umezawa, K.; Mori, M.; Ogawa, T.; et al. ErbB receptor tyrosine kinase/NF-kappaB signaling controls mammosphere formation in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 6584–6589. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Aref, A.R.; Cohoon, T.J.; Barbie, T.U.; Imamura, Y.; Yang, S.; Moody, S.E.; Shen, R.R.; Schinzel, A.C.; Thai, T.C.; et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014, 4, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Gotz, M.E.; Tohidnezhad, M.; Brandenburg, L.O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Yoshida, A.; Kitajima, S.; Li, F.; Cheng, C.; Takegami, Y.; Kohno, S.; Wan, Y.S.; Hayashi, N.; Muranaka, H.; Nishimoto, Y.; et al. MicroRNA-140 mediates RB tumor suppressor function to control stem cell-like activity through interleukin-6. Oncotarget 2017, 8, 13872–13885. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Bhateja, P.; Chiu, M.; Wildey, G.; Lipka, M.B.; Fu, P.; Yang, M.C.L.; Ardeshir-Larijani, F.; Sharma, N.; Dowlati, A. Retinoblastoma mutation predicts poor outcomes in advanced non small cell lung cancer. Cancer Med. 2019, 8, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Taura, M.; Suico, M.A.; Koyama, K.; Komatsu, K.; Miyakita, R.; Matsumoto, C.; Kudo, E.; Kariya, R.; Goto, H.; Kitajima, S.; et al. Rb/E2F1 regulates the innate immune receptor Toll-like receptor 3 in epithelial cells. Mol. Cell. Biol. 2012, 32, 1581–1590. [Google Scholar]
- Zhu, X.; Pattenden, S.; Bremner, R. pRB is required for interferon-gamma-induction of the MHC class II abeta gene. Oncogene 1999, 18, 4940–4947. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell. Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell. 2019, 73, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Terai, H.; Kitajima, S.; Potter, D.S.; Matsui, Y.; Quiceno, L.G.; Chen, T.; Kim, T.J.; Rusan, M.; Thai, T.C.; Piccioni, F.; et al. ER Stress Signaling Promotes the Survival of Cancer "Persister Cells" Tolerant to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018, 78, 1044–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Asahina, H.; Chen, T.; Guo, S.; Quiceno, L.G.; Cavanaugh, J.D.; Merlino, A.A.; Tange, S.; Terai, H.; Kim, J.W.; et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell 2018, 34, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitajima, S.; Li, F.; Takahashi, C. Tumor Milieu Controlled by RB Tumor Suppressor. Int. J. Mol. Sci. 2020, 21, 2450. https://doi.org/10.3390/ijms21072450
Kitajima S, Li F, Takahashi C. Tumor Milieu Controlled by RB Tumor Suppressor. International Journal of Molecular Sciences. 2020; 21(7):2450. https://doi.org/10.3390/ijms21072450
Chicago/Turabian StyleKitajima, Shunsuke, Fengkai Li, and Chiaki Takahashi. 2020. "Tumor Milieu Controlled by RB Tumor Suppressor" International Journal of Molecular Sciences 21, no. 7: 2450. https://doi.org/10.3390/ijms21072450
APA StyleKitajima, S., Li, F., & Takahashi, C. (2020). Tumor Milieu Controlled by RB Tumor Suppressor. International Journal of Molecular Sciences, 21(7), 2450. https://doi.org/10.3390/ijms21072450