Predominant Distribution of the RNAi Machinery at Apical Adherens Junctions in Colonic Epithelia Is Disrupted in Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
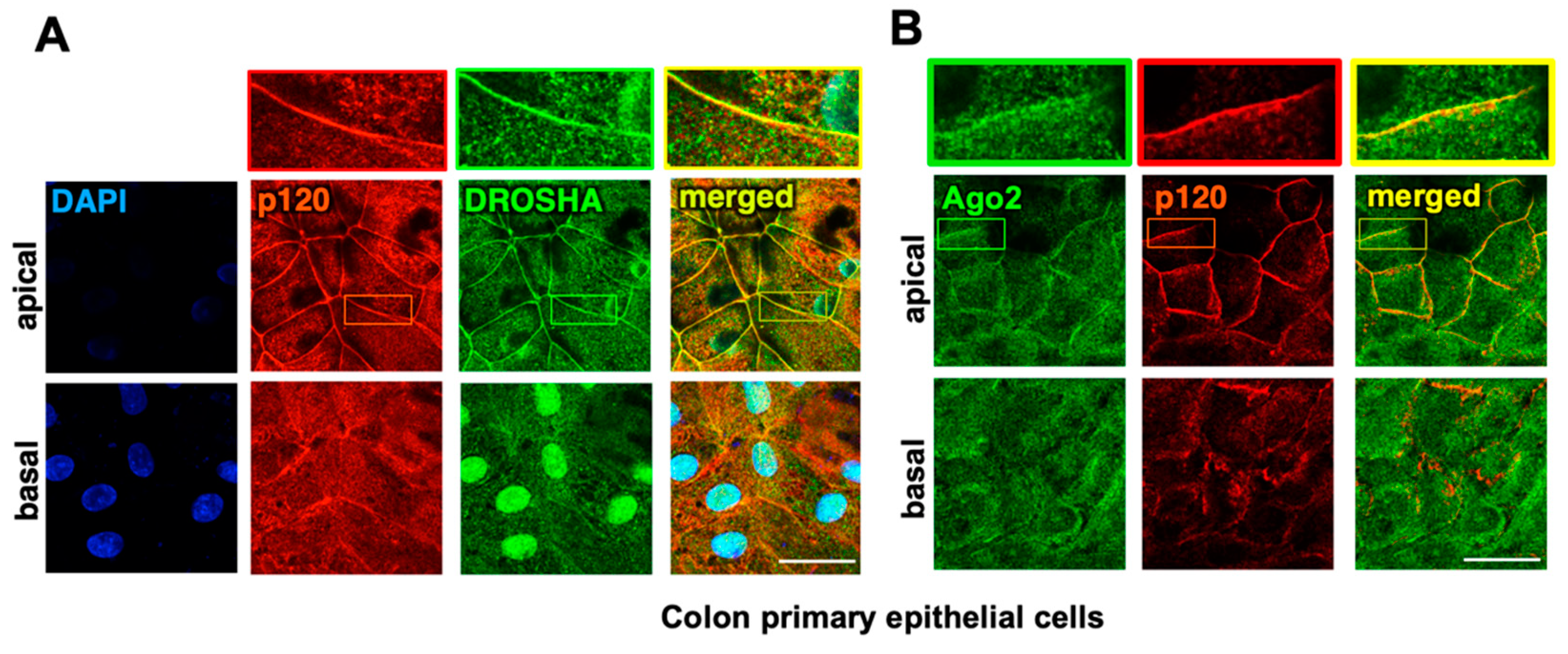
2.1. The Core Components of the RNAi Machinery Localize at Apical Adherens Junctions in Normal Colon Epithelia
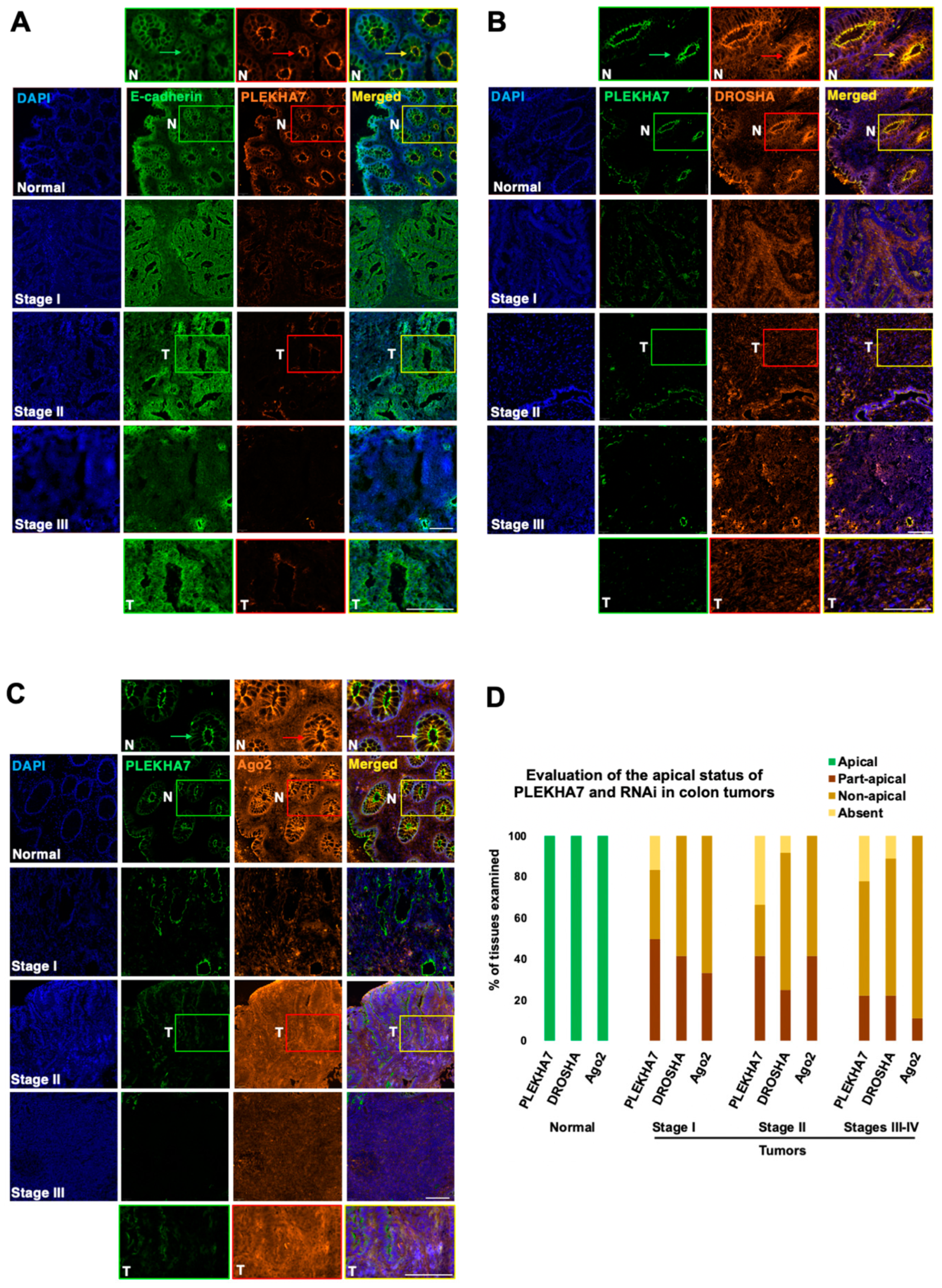
2.2. PLEKHA7 and RNAi Components Are Dysregulated in Human Colon Tumors
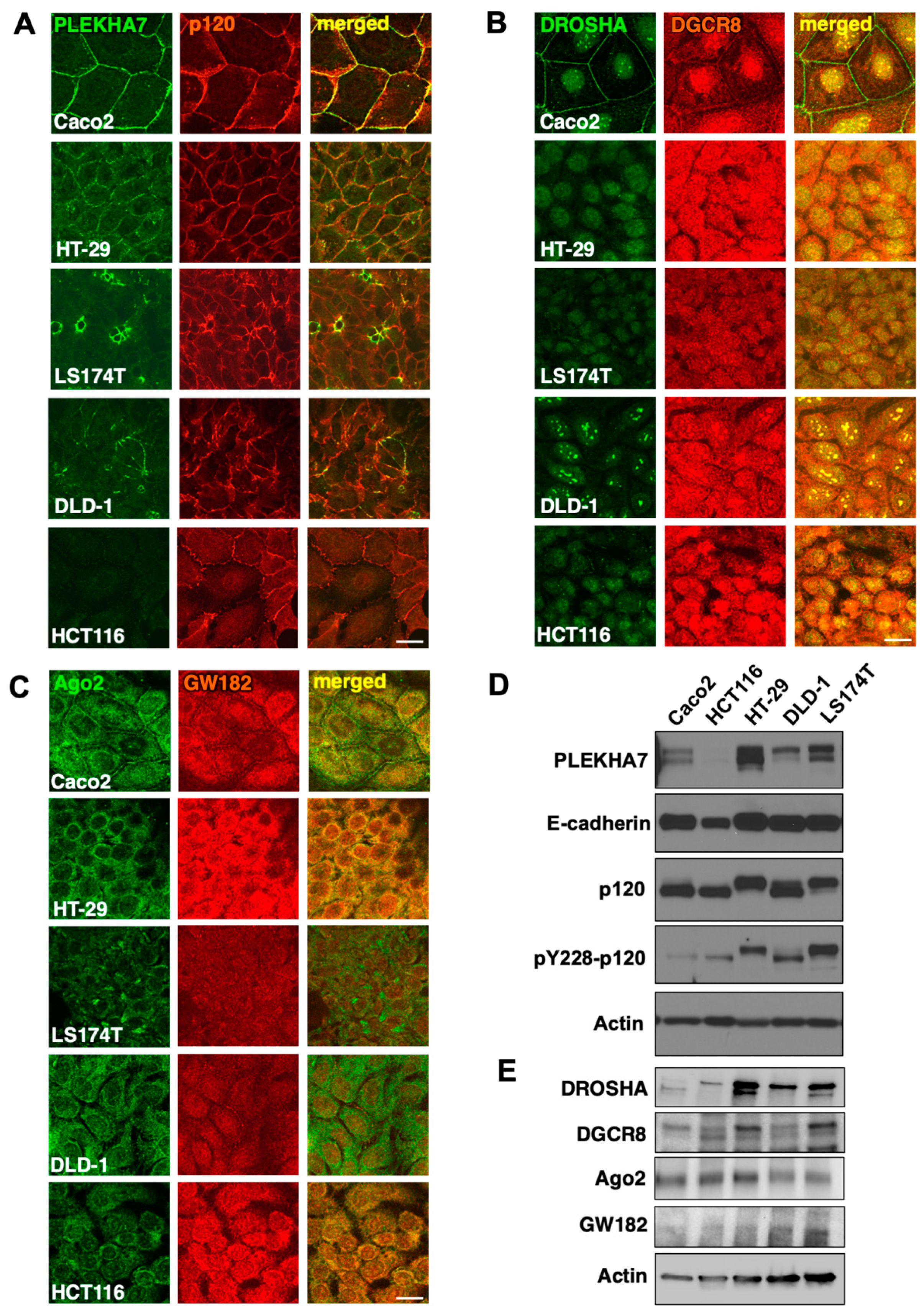
2.3. PLEKHA7 and RNAi Components Are Mis-Localized in Human Colon Cancer Cell Lines
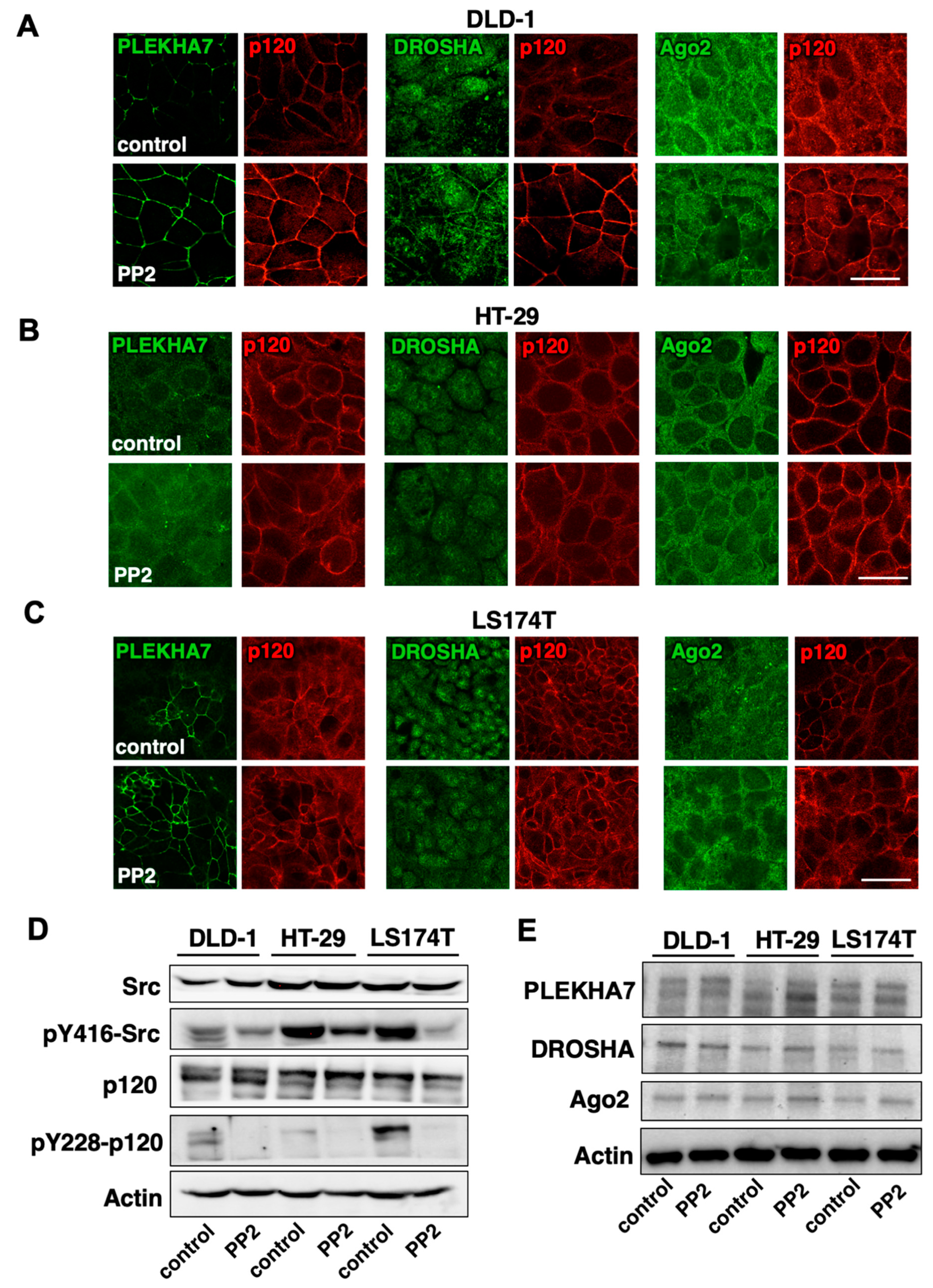
2.4. Src Activity Is Partially Responsible for PLEKHA7-RNAi Mis-Localization in Colon Cancer Cells
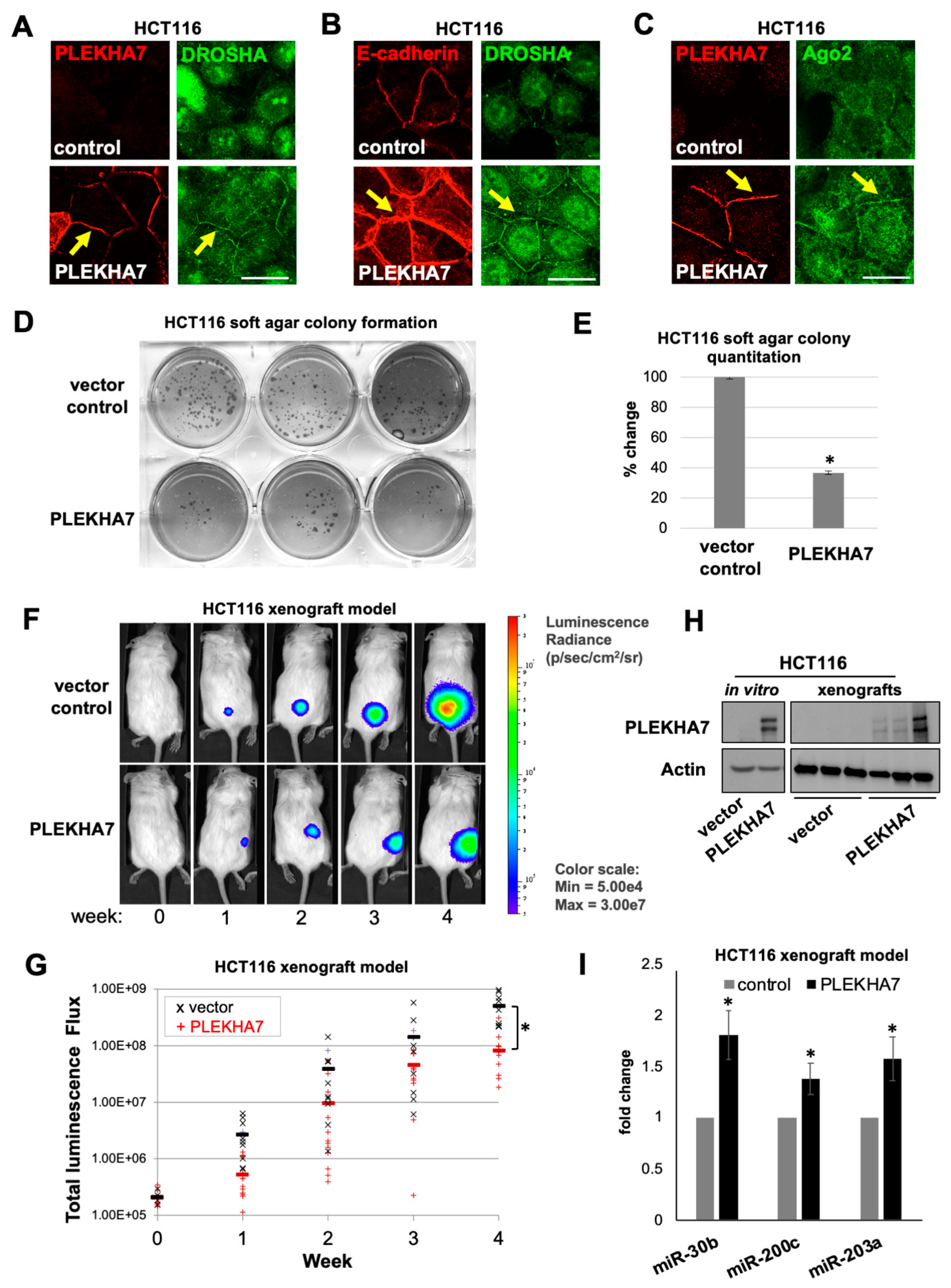
2.5. PLEKHA7 Re-Expression in HCT116 Cells Restores Junctional Localization of RNAi Components and Suppresses Tumor Growth
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Reagents, and Constructs
4.2. Antibodies
4.3. Western Blot
4.4. Cultured Cells Immunofluorescence
4.5. Human Tissue Collection and Immunofluorescence—Immunohistochemistry
4.6. Mouse Xenograft Experiments
4.7. RNA Isolation and qRT-PCR
4.8. Soft Agar Assay
4.9. TCGA Data Analysis
4.10. Statistics and Reproducibility
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ago2 | Argonaute RISC Catalytic Component 2 |
| BLI | Bioluminescence imaging |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DGCR8 | DiGeorge Syndrome Critical Region Gene 8 |
| DROSHA | Drosha Ribonuclease III |
| ECL | Enhanced Chemiluminescence |
| GW182 | Glycine-Tryptophan Protein Of 182 KDa |
| HBSS | Hank’s Balanced Salt Solution |
| HEK293 | Human Embryonic Kidney 293 cells |
| HRP | Horseradish Peroxidase |
| IACUC | Institutional Animal Care and Use Committee |
| LSB | Laemmli Sample Buffer |
| MDCK | Madin-Darby Canine Kidney cells |
| miRNA | microRNA |
| MUSC | Medical University of South Carolina |
| NSG | NOD scid gamma |
| p120 | p120 catenin |
| PBS | Phosphate-Buffered Saline |
| PLEKHA7 | Pleckstrin Homology Domain Containing A7 |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| RIPA | Radioimmunoprecipitation assay buffer |
| RISC | RNA-induced silencing complex |
| RNA | Ribonucleic Acid |
| RNAi | RNA interference |
| TCGA | The Cancer Genome Atlas |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA: A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakradhar, S. Colorectal cancer: 5 big questions. Nature 2015, 521, S16. [Google Scholar] [CrossRef] [PubMed]
- Coskun, M. Intestinal epithelium in inflammatory bowel disease. Front. Med. 2014, 1, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Daulagala, A.C.; Bridges, M.C.; Kourtidis, A. E-cadherin Beyond Structure: A Signaling Hub in Colon Homeostasis and Disease. Int. J. Mol. Sci. 2019, 20, 2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Tepass, U. Adherens junctions: From molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef]
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. p120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 2013, 116, 409–432. [Google Scholar]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef]
- Kourtidis, A.; Ngok, S.P.; Pulimeno, P.; Feathers, R.W.; Carpio, L.R.; Baker, T.R.; Carr, J.M.; Yan, I.K.; Borges, S.; Perez, E.A. Distinct E-cadherin-based complexes regulate cell behaviour through miRNA processing or Src and p120 catenin activity. Nat. Cell Biol. 2015, 17, 1145–1157. [Google Scholar] [CrossRef] [Green Version]
- Dohn, M.R.; Brown, M.V.; Reynolds, A.B. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 2009, 184, 437–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamir, E.R.; Pappalardo, E.; Jorgens, D.M.; Coutinho, K.; Tsai, W.T.; Aziz, K.; Auer, M.; Tran, P.T.; Bader, J.S.; Ewald, A.J. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. J. Cell Biol. 2014, 204, 839–856. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, P.G.; Atlasy, N.; Dieteren, C.E.J.; Navis, A.C.; Venhuizen, J.H.; Veelken, C.; Schubert, D.; Acker-Palmer, A.; Westerman, B.A.; Wurdinger, T. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat. Cell Biol. 2020, 22, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z. E-cadherin’s dark side: Possible role in tumor progression. Biochim. Biophys. Acta 2012, 1826, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Kuphal, S.; Bosserhoff, A.K. E-cadherin cell-cell communication in melanogenesis and during development of malignant melanoma. Arch. Biochem. Biophys. 2012, 524, 43–47. [Google Scholar] [CrossRef]
- Liu, W.F.; Nelson, C.M.; Pirone, D.M.; Chen, C.S. E-cadherin engagement stimulates proliferation via Rac1. J. Cell Biol. 2006, 173, 431–441. [Google Scholar] [CrossRef]
- Meng, W.; Mushika, Y.; Ichii, T.; Takeichi, M. Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell 2008, 135, 948–959. [Google Scholar] [CrossRef] [Green Version]
- Pulimeno, P.; Bauer, C.; Stutz, J.; Citi, S. PLEKHA7 is an adherens junction protein with a tissue distribution and subcellular localization distinct from ZO-1 and E-cadherin. PLoS ONE 2010, 5, e12207. [Google Scholar] [CrossRef]
- Kourtidis, A.; Anastasiadis, P.Z. PLEKHA7 defines an apical junctional complex with cytoskeletal associations and miRNA-mediated growth implications. Cell Cycle 2016, 15, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, K.; Roggiani, F.; De Cecco, L.; Raspagliesi, F.; Carcangiu, M.L.; Nair-Menon, J.; Bagnoli, M.; Bortolomai, I.; Mezzanzanica, D.; Canevari, S. Simultaneous E-cadherin and PLEKHA7 expression negatively affects E-cadherin/EGFR mediated ovarian cancer cell growth. J. Exp. Clin. Cancer Res. 2018, 37, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tille, J.C.; Ho, L.; Shah, J.; Seyde, O.; McKee, T.A.; Citi, S. The Expression of the Zonula Adhaerens Protein PLEKHA7 Is Strongly Decreased in High Grade Ductal and Lobular Breast Carcinomas. PLoS ONE 2015, 10, e0135442. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Anastasiadis, P.Z. Close encounters of the RNAi kind: The silencing life of the adherens junctions. Curr. Opin. Cell Biol. 2018, 54, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Necela, B.; Lin, W.H.; Lu, R.; Feathers, R.W.; Asmann, Y.W.; Thompson, E.A.; Anastasiadis, P.Z. Cadherin complexes recruit mRNAs and RISC to regulate epithelial cell signaling. J. Cell Biol. 2017, 216, 3073–3085. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Reviews. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Grasset, E.; Pinto, M.; Dussaulx, E.; Zweibaum, A.; Desjeux, J.F. Epithelial properties of human colonic carcinoma cell line Caco-2: Electrical parameters. Am. J. Physiol 1984, 247, C260–C267. [Google Scholar] [CrossRef]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Humphries, A.; Wright, N.A. Colonic crypt organization and tumorigenesis. Nat. Rev. 2008, 8, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Ohkubo, T. Tyrosine phosphorylation of p120(ctn) in v-Src transfected L cells depends on its association with E-cadherin and reduces adhesion activity. J. Cell Sci. 2001, 114, 503–512. [Google Scholar] [PubMed]
- Ohkubo, T.; Ozawa, M. The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J. Cell Sci. 2004, 117, 1675–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, S.; Grund, S.E.; Diederichs, S. Alternative splicing affects the subcellular localization of Drosha. Nucleic Acids Res. 2016, 44, 5330–5343. [Google Scholar] [CrossRef]
- Dai, L.; Chen, K.; Youngren, B.; Kulina, J.; Yang, A.; Guo, Z.; Li, J.; Yu, P.; Gu, S. Cytoplasmic Drosha activity generated by alternative splicing. Nucleic Acids Res. 2016, 44, 10454–10466. [Google Scholar] [CrossRef] [Green Version]
- Rajput, A.; Dominguez San Martin, I.; Rose, R.; Beko, A.; Levea, C.; Sharratt, E.; Mazurchuk, R.; Hoffman, R.M.; Brattain, M.G.; Wang, J. Characterization of HCT116 human colon cancer cells in an orthotopic model. J. Surg. Res. 2008, 147, 276–281. [Google Scholar] [CrossRef]
- Joglekar, M.V.; Patil, D.; Joglekar, V.M.; Rao, G.V.; Reddy, D.N.; Mitnala, S.; Shouche, Y.; Hardikar, A.A. The miR-30 family microRNAs confer epithelial phenotype to human pancreatic cells. Islets 2009, 1, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, H.; Liu, J.; Tu, X.; Zang, Y.; Zhu, J.; Chen, J.; Dong, L.; Zhang, J. miR-30 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem. Biophys. Res. Commun. 2012, 417, 1100–1105. [Google Scholar] [CrossRef]
- Mutlu, M.; Raza, U.; Saatci, O.; Eyupoglu, E.; Yurdusev, E.; Sahin, O. miR-200c: A versatile watchdog in cancer progression, EMT, and drug resistance. J. Mol. Med. (Berl) 2016, 94, 629–644. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Das Mahapatra, K.; Pasquali, L.; Crudden, C.; Kular, L.; Akkaya Ulum, Y.Z.; Zhang, L.; Xu Landen, N.; Girnita, L.; Jagodic, M. Genome-Wide Screen for MicroRNAs Reveals a Role for miR-203 in Melanoma Metastasis. J. Investig. Dermatol. 2018, 138, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Chen, W.; Lei, S.; Xiong, L.; Zhao, H.; Liang, D.; Lei, Z.; Zhou, N.; Yao, H.; Liang, Y. Wild-type and mutant p53 differentially modulate miR-124/iASPP feedback following pohotodynamic therapy in human colon cancer cell line. Cell Death Dis. 2017, 8, e3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Gong, L.; Mou, Y.; Han, Y.; Zheng, S. MicroRNA203a3p is a candidate tumor suppressor that targets thrombospondin 2 in colorectal carcinoma. Oncol. Rep. 2019, 42, 1825–1832. [Google Scholar] [PubMed]
- Ogawa, H.; Wu, X.; Kawamoto, K.; Nishida, N.; Konno, M.; Koseki, J.; Matsui, H.; Noguchi, K.; Gotoh, N.; Yamamoto, T. MicroRNAs Induce Epigenetic Reprogramming and Suppress Malignant Phenotypes of Human Colon Cancer Cells. PLoS ONE 2015, 10, e0127119. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.T.; Ye, Y.P.; Zhang, N.J.; Li, T.T.; Wang, S.Y.; Cui, Y.M.; Qi, L.; Wu, P.; Jiao, H.L.; Xie, Y.J. MicroRNA-30b functions as a tumour suppressor in human colorectal cancer by targeting KRAS, PIK3CD and BCL2. J. Pathol. 2014, 232, 415–427. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, Z.; Qin, H.; Gao, Z.; Gao, L. miR-30b regulates migration and invasion of human colorectal cancer via SIX1. Biochem. J. 2014, 460, 117–125. [Google Scholar] [CrossRef]
- Hur, K.; Toiyama, Y.; Takahashi, M.; Balaguer, F.; Nagasaka, T.; Koike, J.; Hemmi, H.; Koi, M.; Boland, C.R.; Goel, A. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 2013, 62, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Karimi Dermani, F.; Amini, R.; Saidijam, M.; Najafi, R. miR-200c, a tumor suppressor that modulate the expression of cancer stem cells markers and epithelial-mesenchymal transition in colorectal cancer. J. Cell. Biochem. 2018, 119, 6288–6295. [Google Scholar] [CrossRef]
- Karimi Dermani, F.; Najafi, R. miR-200c as a Predictive Biomarker for 5-Fluorouracil Chemosensitivity in Colorectal Cancer. J. Gastrointest Cancer 2018, 49, 102–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Z.; Yang, J.; Jiang, B.H.; Di, J.B.; Gao, P.; Peng, L.; Su, X.Q. KIF14 promotes cell proliferation via activation of Akt and is directly targeted by miR-200c in colorectal cancer. Int. J. Oncol. 2018, 53, 1939–1952. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.X.; Yuan, L.; Xue, X.L.; Zhou, M.; Liu, Y.; Zhang, C.; Li, J.P.; Zheng, L.; Hong, M.; Li, X.N. Regulation of colorectal carcinoma stemness, growth, and metastasis by an miR-200c-Sox2-negative feedback loop mechanism. Clin. Cancer Res. 2014, 20, 2631–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Vychytilova-Faltejskova, P.; Svobodova Kovarikova, A.; Grolich, T.; Prochazka, V.; Slaba, K.; Machackova, T.; Halamkova, J.; Svoboda, M.; Kala, Z.; Kiss, I. MicroRNA Biogenesis Pathway Genes Are Deregulated in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4460. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Elfiky, A.; Han, M.; Chen, C.; Saif, M.W. The role of Src in colon cancer and its therapeutic implications. Clin. Colorectal Cancer 2014, 13, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. Is Src the key to understanding metastasis and developing new treatments for colon cancer? Nat. Clin. Pr. Gastroenterol. Hepatol. 2008, 5, 306–307. [Google Scholar] [CrossRef]
- Mariner, D.J.; Anastasiadis, P.; Keilhack, H.; Bohmer, F.D.; Wang, J.; Reynolds, A.B. Identification of Src phosphorylation sites in the catenin p120ctn. J. Biol. Chem. 2001, 276, 28006–28013. [Google Scholar] [CrossRef] [Green Version]
- Bajikar, S.S.; Wang, C.C.; Borten, M.A.; Pereira, E.J.; Atkins, K.A.; Janes, K.A. Tumor-Suppressor Inactivation of GDF11 Occurs by Precursor Sequestration in Triple-Negative Breast Cancer. Dev. Cell 2017, 43, 418–435. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair-Menon, J.; Daulagala, A.C.; Connor, D.M.; Rutledge, L.; Penix, T.; Bridges, M.C.; Wellslager, B.; Spyropoulos, D.D.; Timmers, C.D.; Broome, A.-M.; et al. Predominant Distribution of the RNAi Machinery at Apical Adherens Junctions in Colonic Epithelia Is Disrupted in Cancer. Int. J. Mol. Sci. 2020, 21, 2559. https://doi.org/10.3390/ijms21072559
Nair-Menon J, Daulagala AC, Connor DM, Rutledge L, Penix T, Bridges MC, Wellslager B, Spyropoulos DD, Timmers CD, Broome A-M, et al. Predominant Distribution of the RNAi Machinery at Apical Adherens Junctions in Colonic Epithelia Is Disrupted in Cancer. International Journal of Molecular Sciences. 2020; 21(7):2559. https://doi.org/10.3390/ijms21072559
Chicago/Turabian StyleNair-Menon, Joyce, Amanda C. Daulagala, Dean M. Connor, Lauren Rutledge, Trevor Penix, Mary Catherine Bridges, Bridgette Wellslager, Demetri D. Spyropoulos, Cynthia D. Timmers, Ann-Marie Broome, and et al. 2020. "Predominant Distribution of the RNAi Machinery at Apical Adherens Junctions in Colonic Epithelia Is Disrupted in Cancer" International Journal of Molecular Sciences 21, no. 7: 2559. https://doi.org/10.3390/ijms21072559
APA StyleNair-Menon, J., Daulagala, A. C., Connor, D. M., Rutledge, L., Penix, T., Bridges, M. C., Wellslager, B., Spyropoulos, D. D., Timmers, C. D., Broome, A. -M., & Kourtidis, A. (2020). Predominant Distribution of the RNAi Machinery at Apical Adherens Junctions in Colonic Epithelia Is Disrupted in Cancer. International Journal of Molecular Sciences, 21(7), 2559. https://doi.org/10.3390/ijms21072559