Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Local DAG Signaling Regulation in Cardiomyocytes
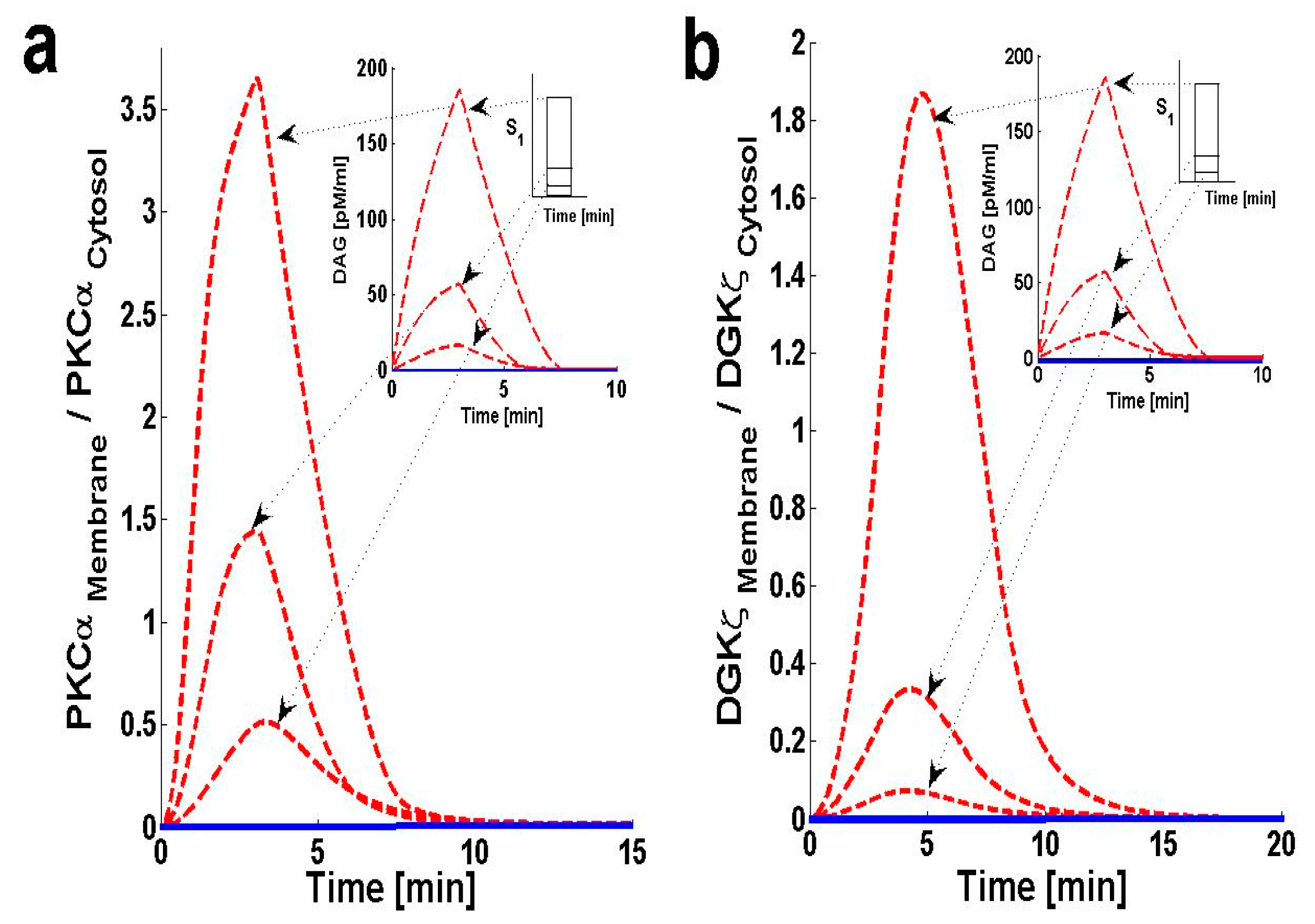
2.2. PKCα and DGKζ Translocation in Cardiomyocytes
2.3. Effects of DGKζ Overexpression on the Translocation Characteristics of PKCα in Cardiomyocytes
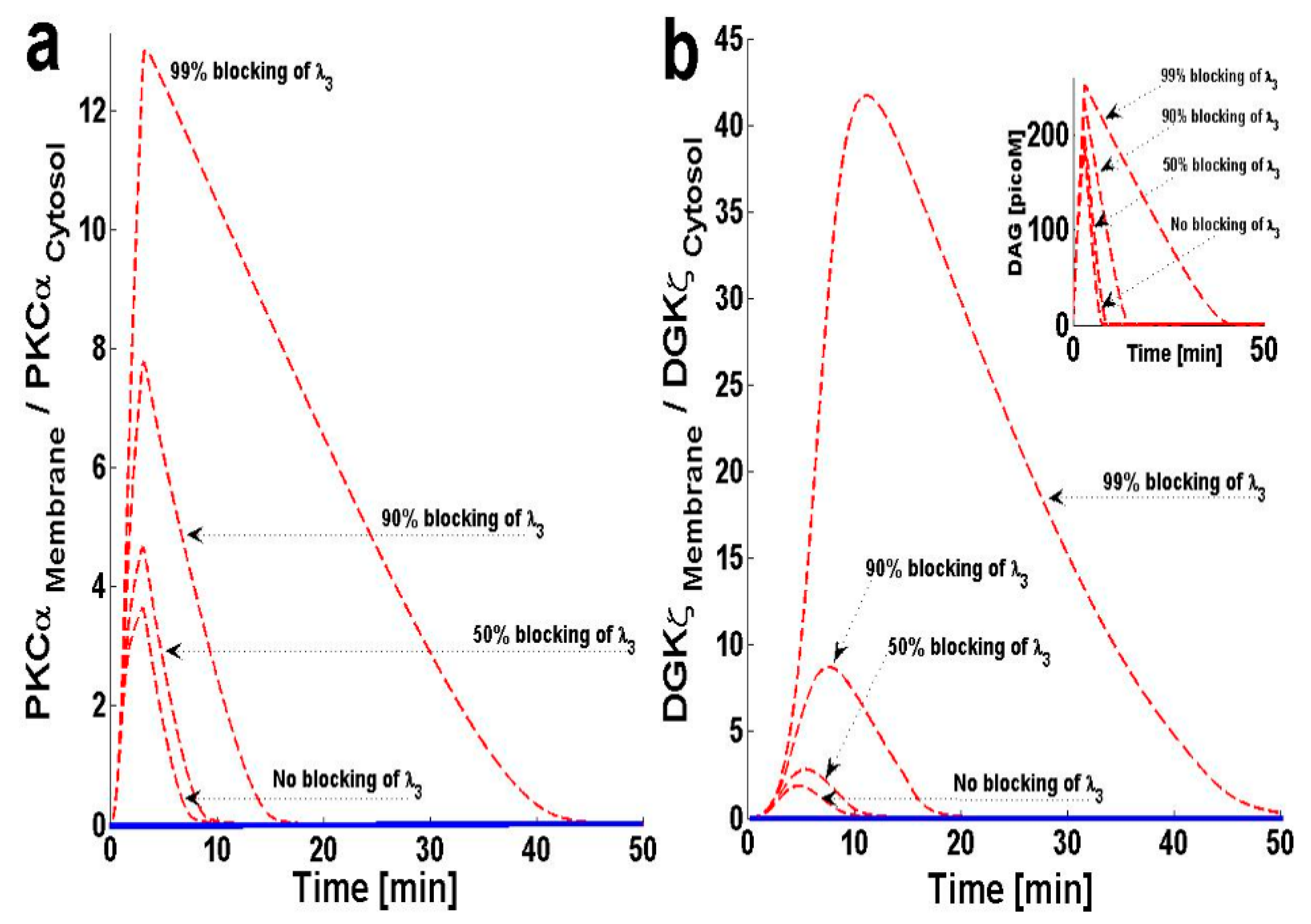
2.4. Effects of PKCIαActive’s Translocation Parameter λ3 on DAG–PKCα Signaling Characteristics
2.5. Effects of PKCIIαActive’s Deactivation Parameter k0 on DAG–PKCα Signaling Characteristics
2.6. Effect of Forward Rate Constant, ‘k2’, on DAG–PKCα Signaling Characteristics
3. Discussion
4. Materials and Methods
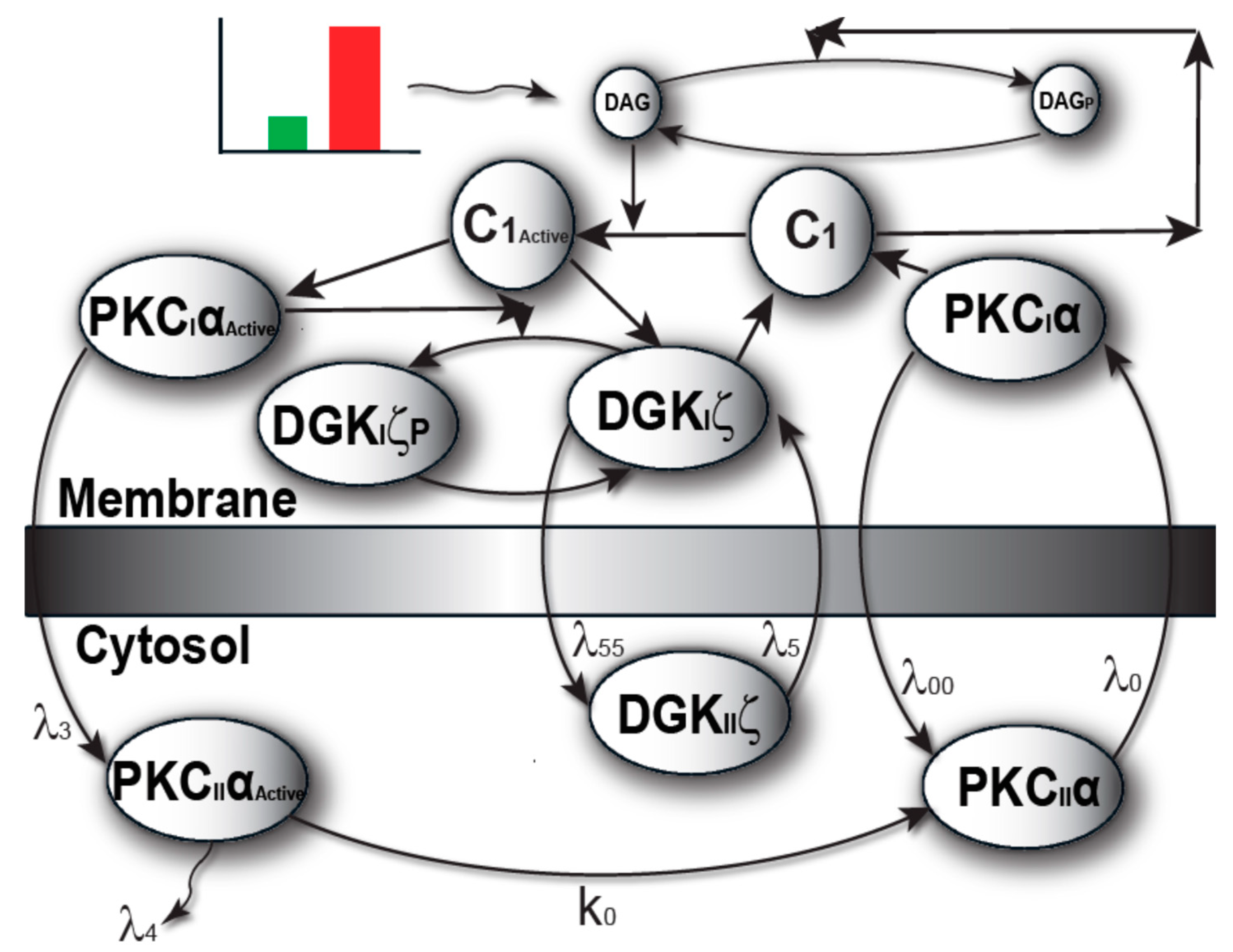
4.1. Biochemical Reactions
4.2. Induction
4.3. Temporal Dynamics
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Ertl, G.; Gaudron, P.; Neubauer, S.; Bauer, B.; Horn, M.; Hu, K.; Tian, R. Cardiac dysfunction and development of heart failure. Eur. Heart J. 1993, 14 (Suppl. A), 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2009, 81, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwinger, R.H.; Böhm, M.; Müller-Ehmsen, J.; Uhlmann, R.; Schmidt, U.; Stäblein, A.; Uberfuhr, P.; Kreuzer, E.; Reichart, B.; Eissner, H.J. Effect of inotropic stimulation on the negative force-frequency relationship in the failing human heart. Circulation 1993, 88, 2267–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, J.; Abriel, H. Neurohormonal regulation of cardiac ion channels in chronic heart failure. J. Cardiovasc. Pharm. 2009, 54, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Molkentin, J. Molecular Pathways Underlying Cardiac Remodeling During Pathophysiological Stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Anderson, M.E. Mechanisms of Altered Ca2+ Handling in Heart Failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [Green Version]
- Jarkko, P. Regulation of Cardiac Responses to Increased Load. Role of Endothelin-1, Angiotensin II and Collagen XV; Department of Pharmacology and Toxicology and Biocenter Oulu, University of Oulu, P.O.Box 5000, FIN-90014 University of Oulu, Finland: Oulu, Finland, 2002. [Google Scholar]
- Liu, Q.; Chen, X.; Macdonnell, S.M.; Kranias, E.G.; Lorenz, J.N.; Leitges, M.; Houser, S.R.; Molkentin, J.D. Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ. Res. 2009, 105, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Michael, H.; Harvey, H.; Sven, T.P.; Matthew, C.K.; Raisa, K.; Andrew, N.C.; Thomas, F.K.; Timothy, E.H.; Gerald, W.D., II; Walter, J.K.; et al. Pharmacological- and Gene Therapy-Based Inhibition of Protein Kinase Cα/β Enhances Cardiac Contractility and Attenuates Heart Failure. Circulation 2006, 114, 574–582. [Google Scholar]
- Hambleton, M.; York, A.; Sargent, M.A.; Kaiser, R.A.; Lorenz, J.N.; Robbins, J.; Molkentin, J.D. Inducible and myocyte-specific inhibition of PKCalpha enhances cardiac contractility and protects against infarction-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3768–H3771. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.F. Cardiac Actions of Protein Kinase C Isoforms. Physiology 2012, 27, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Hahn, H.S.; Marreez, Y.; Odley, A.; Sterbling, A.; Yussman, M.G.; Hilty, K.C.; Bodi, I.; Liggett, S.B.; Schwartz, A.; Dorn, G.W. Protein kinase C alpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circ. Res. 2003, 93, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, T.; Takeishi, Y.; Takahashi, H.; Shishido, T.; Niizeki, T.; Koyama, Y.; Shiga, R.; Nozaki, N.; Nakajima, O.; Nishimaru, K.; et al. Cardiac-specific overexpression of diacylglycerol kinase zeta prevents Gq protein-coupled receptor agonist-induced cardiac hypertrophy in transgenic mice. Circulation 2006, 113, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niizeki, T.; Takeishi, Y.; Arimoto, T.; Takahashi, H.; Shishido, T.; Koyama, Y.; Goto, K.; Walsh, R.A.; Kubota, I. Cardiac-specific overexpression of diacylglycerol kinase zeta attenuates left ventricular remodeling and improves survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1105–H1112. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Takeishi, Y.; Arimoto, T.; Niizeki, T.; Kitahara, T.; Goto, K.; Walsh, R.A.; Kubota, I. Diacylglycerol kinase zeta attenuates pressure overload-induced cardiac hypertrophy. Circ. J. 2007, 71, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Niizeki, T.; Takeishi, Y.; Kitahara, T.; Arimoto, T.; Ishino, M.; Bilim, O.; Suzuki, S.; Sasaki, T.; Nakajima, O.; Walsh, R.A.; et al. Diacylglycerol kinase-epsilon restores cardiac dysfunction under chronic pressure overload: A new specific regulator of Galpha(q) signaling cascade. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H245–H255. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Debra, S.R.; Stephen, M.P.; Matthew, K.T. Diacylglycerol kinases. Cell. Signal. 2004, 16, 983–989. [Google Scholar]
- Takeishi, Y.; Goto, K.; Kubota, I. Role of diacylglycerol kinase in cellular regulatory processes: A new regulator for cardiomyocyte hypertrophy. Pharm. Ther. 2007, 115, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Prescott, S.M.; Topham, M.K. Association of diacylglycerol kinase zeta with protein kinase C alpha: Spatial regulation of diacylglycerol signaling. J. Cell Biol. 2003, 160, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Gharbi, S.I.; Rincón, E.; Avila-Flores, A.; Torres-Ayuso, P.; Almena, M.; Cobos, M.A.; Albar, J.P.; Mérida, I. Diacylglycerol kinase ζ controls diacylglycerol metabolism at the immunological synapse. Mol. Biol. Cell 2011, 22, 4406–4414. [Google Scholar] [CrossRef]
- Luo, B.; Prescott, S.M.; Topham, M.K. Protein Kinase C{alpha} Phosphorylates and Negatively Regulates Diacylglycerol Kinase {zeta}. J. Biol. Chem. 2003, 278, 39542–39547. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Brognard, J.; Newton, A.C. The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 2008, 283, 6300–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, G.W.I.I.; Molkentin, J.D. Manipulating cardiac contractility in heart failure: Data from mice and men. Circulation 2004, 109, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S.; Tang, W.H. Pathophysiology of congestive heart failure. Rev. Cardiovasc. Med. 2003, 4, S14–S20. [Google Scholar]
- Houser, S.R.; Margulies, K.B. Is depressed myocyte contractility centrally involved in heart failure? Circ. Res. 2003, 92, 350–358. [Google Scholar] [CrossRef]
- Colucci, W.S.; Leatherman, G.F.; Ludmer, P.L.; Gauthier, D.F. Adrenergic inotropic responsiveness of patients with heart failure: Studies with intracoronary dobutamine infusion. Circ. Res. 1987, 61 (Suppl. I), I82–I86. [Google Scholar]
- Feldman, A.M.; Bristow, M.R.; Parmley, W.W.; Carson, P.E.; Pepine, C.J.; Gilbert, E.M.; Strobeck, J.E.; Hendrix, G.H.; Powers, E.R.; Bain, R.P.; et al. Effects of vesnarinone on morbidity and mortality in patients with heart failure. Vesnarinone Study Group. N. Engl. J. Med. 1993, 329, 149–155. [Google Scholar] [CrossRef]
- Chatterjee, K.; De Marco, T. Role of nonglycosidic inotropic agents: Indications, ethics, and limitations. Med. Clin. N. Am. 2003, 87, 391–418. [Google Scholar] [CrossRef]
- Warner-Stevenson, L. Clinical use of inotropic therapy for heart failure: Looking backward or forward? Circulation 2003, 108, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Felker, G.M.; O’Connor, C.M. Inotropic therapy for heart failure: An evidence-based approach. Am. Heart J. 2001, 142, 393–401. [Google Scholar] [CrossRef]
- Packer, M.; Gheorghiade, M.Y.; James, B.; Costantini, P.J.; Adams, K.F.; Cody, R.J.; Smith, L.K.; Van Voorhees, L.; Gourley, L.A.; Jolly, M.K.; et al. Withdrawal of digoxin from patients with chronic heart failure treated with angiotensin-converting- enzyme inhibitors. Radiance Study. N. Engl. J. Med. 1993, 329, 1–7. [Google Scholar] [CrossRef]
- Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. The Digitalis Investigation Group. N. Engl. J. Med. 1993, 336, 525–533. [Google Scholar]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zaho, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef]
- Pi, Y.; Walker, J.W. Diacylglycerol and fatty acids synergistically increase cardiomyocyte contraction via activation of PKC. Am. J. Phsiol. Heart Cir. Physiol. 2000, 279, H26–H34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosatos, K.; Bharadwaj, K.G.; Lymperopoulos, A.; Ikeda, S.; Khan, R.; Hu, Y.; Agarwal, R.; Yu, S.; Jiang, H.; Steinberg, S.F.; et al. Cardiomyocyte lipids impair β-adrenergic receptor function via PKC activation. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E489–E499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borradaile, N.M.; Buhman, K.K.; Listenberger, L.L.; Magee, C.J.; Morimoto, E.T.; Ory, D.S.; Schaffer, J.E. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol. Biol. Cell 2006, 17, 770–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.D. Fatty acyl CoA-mediated inhibition of endoplasmic reticulum assembly. Biochim. Biophys. Acta 2004, 1693, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopee, N.V.; Sharma, R.P. Selective and transient activation of protein kinase C alpha by fumonisin B1, a ceramide synthase inhibitor mycotoxin, in cultured porcine renal cells. Life Sci. 2004, 74, 1541–1559. [Google Scholar] [CrossRef] [PubMed]
- Abramovici, H.; Hogan, A.B.; Obagi, C.; Topham, M.K.; Gee, S.H. Diacylglycerol kinase-zeta localization in skeletal muscle is regulated by phosphorylation and interaction with syntrophins. Mol. Biol. Cell 2003, 14, 4499–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharbi, S.I.; Avila-Flores, A.; Soutar, D.; Orive, A.; Koretzky, G.A.; Albar, J.P.; Merida, I. Transient PKCα shuttling to the immunological synapse is governed by Diacylglycerol kinase ζ and regulates L-selection shedding. J. Cell Sci. 2013, 126, 2176–2186. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Kagaya, Y.; Takahashi, J.; Sugie, T.; Ohta, J.; Watanabe, J.; Shirato, K.; Kondo, H.; Goto, K. Gene Expression and In Situ Localization of Diacylglycerol Kinase Isozymes in Normal and Infarcted Rat Hearts Effects of Captopril Treatment. Circ. Res. 2001, 89, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Sabourin, J.; Antigny, F.; Robin, E.; Frieden, M.; Raddatz, E. Activation of Transient Receptor Potential Canonical 3 (TRPC3)-mediated Ca+2 Entry by A1 Adenosine Receptor in Cardiomyocytes Disturbs Atrioventricular Conduction. J. Biol. Chem. 2012, 287, 26688–26701. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J.; Izumo, S. Signal Transduction Pathways of Angiotensin-II Induced c-fos Gene Expression in Cardiac Myocytes In-Vitro Roles of Phospholipid-Derived Second Messengers. Circ. Res. 1993, 73, 424–438. [Google Scholar] [CrossRef] [Green Version]
- Topham, M.K.; Bunting, M.; Zimmerman, G.A.; McIntyre, T.M.; Blackshear, P.J.; Prescott, S.M. Protein kinase C regulates the nuclear localization of diacylglycerol kinase-Zeta. Nature 1998, 394, 697–700. [Google Scholar] [CrossRef]
- Goto, K.; Kondo, H. A 104-kDa diacylglycerol kinase containing ankyrin-like repeats localizes in the cell nucleus. Proc. Natl. Acad. Sci. USA 1996, 93, 11196–11201. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, N. Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism. Int. J. Mol. Sci. 2020, 21, 2561. https://doi.org/10.3390/ijms21072561
Aslam N. Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism. International Journal of Molecular Sciences. 2020; 21(7):2561. https://doi.org/10.3390/ijms21072561
Chicago/Turabian StyleAslam, Naveed. 2020. "Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism" International Journal of Molecular Sciences 21, no. 7: 2561. https://doi.org/10.3390/ijms21072561
APA StyleAslam, N. (2020). Increase in PKCα Activity during Heart Failure Despite the Stimulation of PKCα Braking Mechanism. International Journal of Molecular Sciences, 21(7), 2561. https://doi.org/10.3390/ijms21072561