Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
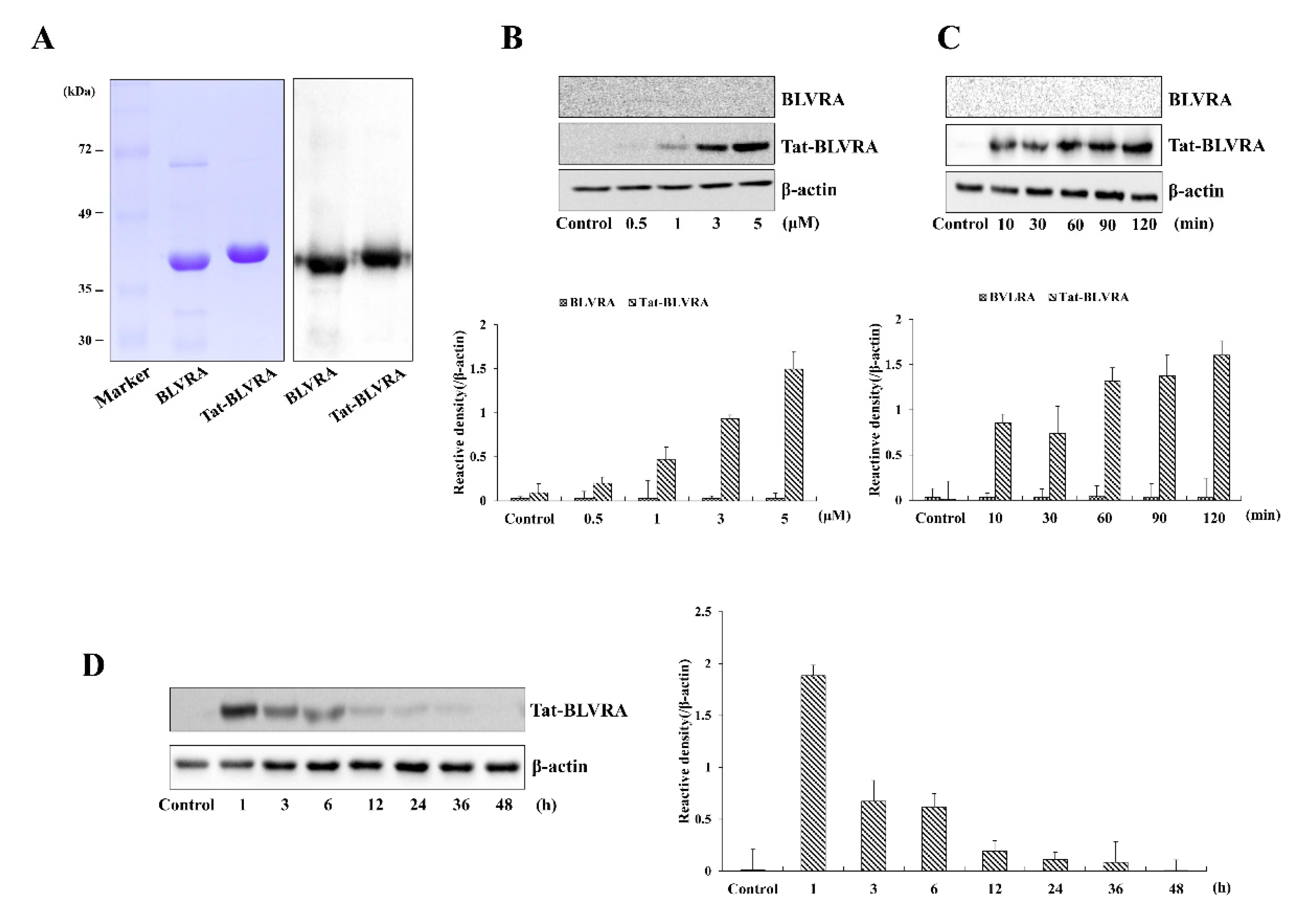
2.1. Purification and Transduction of Tat-BLVRA into HT-22 Cells
2.2. Effect of Tat-BLVRA against H2O2-Induced Cell Death
2.3. Protective Effect of Tat-BLVRA against H2O2-Induced Cytotoxicity
2.4. Effects of Tat-BLVRA on H2O2-Induced Activation of MAPKs and Apoptosis
2.5. Effects of Tat-BLVRA on Ischemic Insults
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Viability Measurements
4.2. Transduction of Tat-BLVRA into HT-22 Cells
4.3. Western Blot Analysis
4.4. Measurement of Reactive Oxygen Species (ROS) and DNA Fragmentation
4.5. Measurement of Activation of Akt and MAPK as well as Apoptosis Signals
4.6. Experimental Animals
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Maines, M.D. New Insights into Biliverdin Reductase Functions: Linking Heme Metabolism to Cell Signaling. Physiology 2005, 20, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komuro, A.; Tobe, T.; Nakano, Y.; Yamaguchi, T.; Tomita, M. Cloning and characterization of the cDNA encoding human biliverdin-IXα reductase. Biochim. Biophys. Acta Gene Struct. Expr. 1996, 1309, 89–99. [Google Scholar] [CrossRef]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barañano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florczyk, U.M.; Jozkowicz, A.; Dulak, J. Biliverdin reductase: New features of an old enzyme and its potential therapeutic significance. Pharmacol. Rep. 2008, 60, 38–48. [Google Scholar] [PubMed]
- Gibbs, P.E.M.; Miralem, T.; Maines, M.D. Biliverdin reductase: A target for cancer therapy? Front. Pharmacol. 2015, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, C. Bilirubin and brain: A pharmacological approach. Neuropharmacology 2017, 118, 113–123. [Google Scholar] [CrossRef]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2018, 56, 2922–2943. [Google Scholar] [CrossRef]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- Doré, S.; Takahashi, M.; Ferris, C.D.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Boil. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Tetzlaff, W.; Paty, N.W.; Cynader, M.S. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Boil. Med. 2006, 40, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Marmarosh, N.; Shen, J.; Torno, M.D.; Kravets, A.; Hu, Z.; Maines, M.D. Human biliverdin reductase: A member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 7109–7114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, R.; Yao, Y.; Han, M.; Zhao, X.; Liu, X.-C.; Wei, J.; Luo, Y.; Zhang, J.; Zhou, J.; Wang, S.; et al. Biliverdin reductase mediates hypoxia-induced EMT via PI3-kinase and Akt. J. Am. Soc. Nephrol. 2008, 19, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, B.; Otterbein, L.E. Go Green: The Anti-Inflammatory Effects of Biliverdin Reductase. Front. Pharmacol. 2012, 3, 47. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.T.; MacManus, J.P. Molecular mechanisms of cerebral ischemia-induced neuronal death. Adv. Appl. Microbiol. 2002, 221, 93–148. [Google Scholar]
- Frantseva, M.V.; Carlen, P.; Velazquez, J.L.P. Dynamics of intracellular calcium and free radical production during ischemia in pyramidal neurons. Free Radic. Boil. Med. 2001, 31, 1216–1227. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive Oxygen Radicals in Signaling and Damage in the Ischemic Brain. Br. J. Pharmacol. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Maines, M.D. Potential application of biliverdin reductase and its fragments to modulate insulin/IGF-1/MAPK/PI3-K signaling pathways in therapeutic settings. Curr. Drug Targets 2010, 11, 1586–1594. [Google Scholar] [CrossRef]
- Gibbs, P.E.M.; Tudor, C.; Maines, M.D. Biliverdin Reductase: More than a Namesake – The Reductase, Its Peptide Fragments, and Biliverdin Regulate Activity of the Three Classes of Protein Kinase C. Front. Pharmacol. 2012, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Lerner-Marmarosh, N.; Miralem, T.; Gibbs, P.E.M.; Maines, M.D. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Kim, D.W.; Jo, H.S.; Cho, S.B.; Park, J.H.; Lee, C.H.; Choi, Y.J.; Park, S.Y.; Kim, S.T.; Yu, Y.H.; et al. Tat-biliverdin reductase A inhibits inflammatory response by regulation of MAPK and NF-ĸB pathway in Raw 264.7 cells and edema mouse model. Mol. Immunol. 2015, 63, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Enokido, Y.; Aoshima, H.; Uchiyama, Y.; Hatanaka, H. Changes in mitochondrial membrane potential during oxidative stress-induced apoptosis in PC12 cells. J. Neurosci. Res. 1997, 50, 413–420. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.; Liang, J. Activation of the Nrf2 defense pathway contributes to neuroprotective effects of phloretin on oxidative stress injury after cerebral ischemia/reperfusion in rats. J. Neurol. Sci. 2015, 351, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Wadia, J.S.; Dowdy, S.F. Protein transduction technology. Curr. Opin. Biotechnol. 2002, 13, 52–56. [Google Scholar] [CrossRef]
- Moon, J.-I.; Han, M.-J.; Yu, S.-H.; Lee, E.-H.; Kim, S.-M.; Han, K.; Park, C.-H.; Kim, C.-H. Enhanced delivery of protein fused to cell penetrating peptides to mammalian cells. BMB Rep. 2019, 52, 324–329. [Google Scholar] [CrossRef]
- Kubo, E.; Fatma, N.; Akagi, Y.; Beier, D.R.; Singh, S.P.; Singh, D.P. TAT-mediated PRDX6 protein transduction protects against eye lens epithelial cell death and delays lens opacity. Am. J. Physiol. Physiol. 2008, 294, C842–C855. [Google Scholar] [CrossRef] [Green Version]
- Sakurazawa, M.; Katsura, K.; Saito, M.; Asoh, S.; Ohta, S.; Katayama, Y. Mild hypothermia enhanced the protective effect of protein therapy with transductive anti-death FNK protein using a rat focal transient cerebral ischemia model. Brain Res. 2012, 1430, 86–92. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.J.; Kim, D.W.; Lee, Y.P.; Ahn, E.H.; Jo, H.S.; Kim, D.-S.; Kwon, O.-S.; Kang, T.-C.; Cho, Y.-J.; Park, J.; et al. Tat-glyoxalase protein inhibits against ischemic neuronal cell damage and ameliorates ischemic injury. Free Radic. Boil. Med. 2014, 67, 195–210. [Google Scholar] [CrossRef]
- Yeo, H.J.; Shin, H.J.Y.M.J.; You, J.H.; Kim, J.S.; Kim, M.Y.; Kim, D.W.; Kim, D.-S.; Eum, W.S.; Choi, S.Y. Transduced Tat-CIAPIN1 reduces the inflammatory response on LPS- and TPA-induced damages. BMB Rep. 2019, 52, 695–699. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.J.; Yoo, D.Y.; Kim, D.W.; Yeo, H.J.; Bin Cho, S.; Hyeon, J.; Park, J.H.; Park, J.; Eum, W.S.; Hwang, H.S.; et al. Neuroprotective effect of PEP-1-peroxiredoxin2 on CA1 regions in the hippocampus against ischemic insult. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.J.; Shin, M.J.; Yeo, E.J.; Choi, Y.J.; Kim, D.W.; Kim, D.-S.; Eum, W.S.; Choi, S.Y. Tat-CIAPIN1 inhibits hippocampal neuronal cell damage through the MAPK and apoptotic signaling pathways. Fre. Radic. Boil. Med. 2019, 135, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, M.; Kim, D.W.; Shin, M.J.; Son, O.; Jo, H.S.; Yeo, H.J.; Bin Cho, S.; Park, J.H.; Lee, C.H.; et al. Transduced PEP-1-PON1 proteins regulate microglial activation and dopaminergic neuronal death in a Parkinson’s disease model. Biomaterials 2015, 64, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.J.; Yeo, E.J.; Shin, M.J.; Choi, Y.J.; Lee, C.H.; Kwon, H.Y.; Kim, D.W.; Eum, W.S.; Choi, S.Y. Protective effects of Tat-DJ-1 protein against streptozotocin-induced diabetes in a mice model. BMB Rep. 2018, 51, 362–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Blanco, R.; Rivera, R.; Ballabriga, J.; Pozas, E. Bcl-2, Bax, and Bcl-x expression in the CA1 area of the hippocampus following transient forebrain ischemia in the adult gerbil. Exp. Brain Res. 1998, 121, 167–173. [Google Scholar] [CrossRef]
- Park, S.; Kim, J.A.; Choi, S.; Suh, S.H. Superoxide is a potential culprit of caspase-3 dependent endothelial cell death induced by lysophosphatidylcholine. J. Physiol. Pharmacol. 2010, 61, 375–381. [Google Scholar]
- Pachori, A.S.; Smith, A.; McDonald, P.; Zhang, L.; Dzau, V.J.; Melo, L.G. Heme-oxygenase-1-induced protection against hypoxia/reoxy genation is dependent on biliverdin reductase and its interaction with PI3K/Akt pathway. J. Mol. Cell. Cardiol. 2007, 43, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Busserolles, J.; Megías, J.; Terencio, M.C.; Alcaraz, M.J. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int. J. Biochem. Cell Boil. 2006, 38, 1510–1517. [Google Scholar] [CrossRef]
- Alvarez-Buylla, A.; Ling, C.-Y.; Kirn, J.R. Cresyl violet: A red fluorescent Nissl stain. J. Neurosci. Methods 1990, 33, 129–133. [Google Scholar] [CrossRef]
- Schmued, L.C.; Hopkins, K.J. Flouro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Tanaka, K.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 2001, 32, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wang, S.; Ma, J.; Yao, L.; Xing, H.; Zhang, L.; Liao, L.; Zhu, D. Biliverdin reductase/bilirubin mediates the anti-apoptotic effect of hypoxia in pulmonary arterial smooth muscle cells through ERK1/2 pathway. Exp. Cell Res. 2013, 319, 1973–1987. [Google Scholar] [CrossRef]
- Salido, M.; Gonzalez, J.L.; Vilches, J. Loss of mitochondrial membrane potential is inhibited by bombesin in etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol. Cancer Ther. 2007, 6, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Miralem, T.; Hu, Z.; Torno, M.D.; Lelli, K.M.; Maines, M.D. Small Interference RNA-mediated Gene Silencing of Human Biliverdin Reductase, but Not That of Heme Oxygenase-1, Attenuates Arsenite-mediated Induction of the Oxygenase and Increases Apoptosis in 293A Kidney Cells. J. Boil. Chem. 2005, 280, 17084–17092. [Google Scholar] [CrossRef] [Green Version]
- Philip, L.; Shivakumar, K. cIAP-2 protects cardiac fibroblasts from oxidative damage: An obligate regulatory role for ERK1/2 MAPK and NF-κB. J. Mol. Cell. Cardiol. 2013, 62, 217–226. [Google Scholar] [CrossRef]
- Post, A.; Holsboer, F.; Behl, C. Induction of NF-κB Activity during Haloperidol-Induced Oxidative Toxicity in Clonal Hippocampal Cells: Suppression of NF-κB and Neuroprotection by Antioxidants. J. Neurosci. 1998, 18, 8236–8246. [Google Scholar] [CrossRef] [Green Version]
- Crossthwaite, A.J.; Hasan, S.; Williams, R.J. Hydrogen peroxide-mediated phosphorylation of ERK1/2, Akt/PKB and JNK in cortical neurons: Dependence on Ca2+ and PI3-kinase. J. Neurochem. 2002, 80, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Tanaka, S.; Oikawa, T.; Koike, T.; Takahashi, A.; Itoyama, Y. Expression of microglial response factor-1 in microglia and macrophages following cerebral ischemia in the rat. Brain Res. 2000, 882, 206–211. [Google Scholar] [CrossRef]
- Cheung, W.M.; Wang, C.K.; Kuo, J.S.; Lin, T.N. Changes in the level of glial fibrillary acidic protein (GFAP) after mild and severe focal cerebral ischemia. Chin. J. Physiol. 1999, 42, 227–235. [Google Scholar] [PubMed]
- Kim, S.M.; Ha, J.S.; Han, A.R.; Cho, S.W.; Yang, S.J. Effects of α-lipoic acid on LPS-induced neuroinflammation and NLRP3 inflammasome activation through the regulation of BV-2 microglia cells activation. BMB Rep. 2019, 52, 613–618. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.J.; Shin, M.J.; Kim, D.W.; Yeo, H.J.; Yeo, E.J.; Choi, Y.J.; Sohn, E.J.; Han, K.H.; Park, J.; Lee, K.W.; et al. Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling. Int. J. Mol. Sci. 2020, 21, 2672. https://doi.org/10.3390/ijms21082672
Kim SJ, Shin MJ, Kim DW, Yeo HJ, Yeo EJ, Choi YJ, Sohn EJ, Han KH, Park J, Lee KW, et al. Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling. International Journal of Molecular Sciences. 2020; 21(8):2672. https://doi.org/10.3390/ijms21082672
Chicago/Turabian StyleKim, Sang Jin, Min Jea Shin, Dae Won Kim, Hyeon Ji Yeo, Eun Ji Yeo, Yeon Joo Choi, Eun Jeong Sohn, Kyu Hyung Han, Jinseu Park, Keun Wook Lee, and et al. 2020. "Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling" International Journal of Molecular Sciences 21, no. 8: 2672. https://doi.org/10.3390/ijms21082672
APA StyleKim, S. J., Shin, M. J., Kim, D. W., Yeo, H. J., Yeo, E. J., Choi, Y. J., Sohn, E. J., Han, K. H., Park, J., Lee, K. W., Park, J. K., Cho, Y. -J., Kim, D. -S., Eum, W. S., & Choi, S. Y. (2020). Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling. International Journal of Molecular Sciences, 21(8), 2672. https://doi.org/10.3390/ijms21082672