Identification of the Key Molecular Drivers of Phosphorus Utilization Based on Host miRNA-mRNA and Gut Microbiome Interactions

 , , , , , , , and
, , , , , , , and 
Abstract
:1. Introduction
2. Results
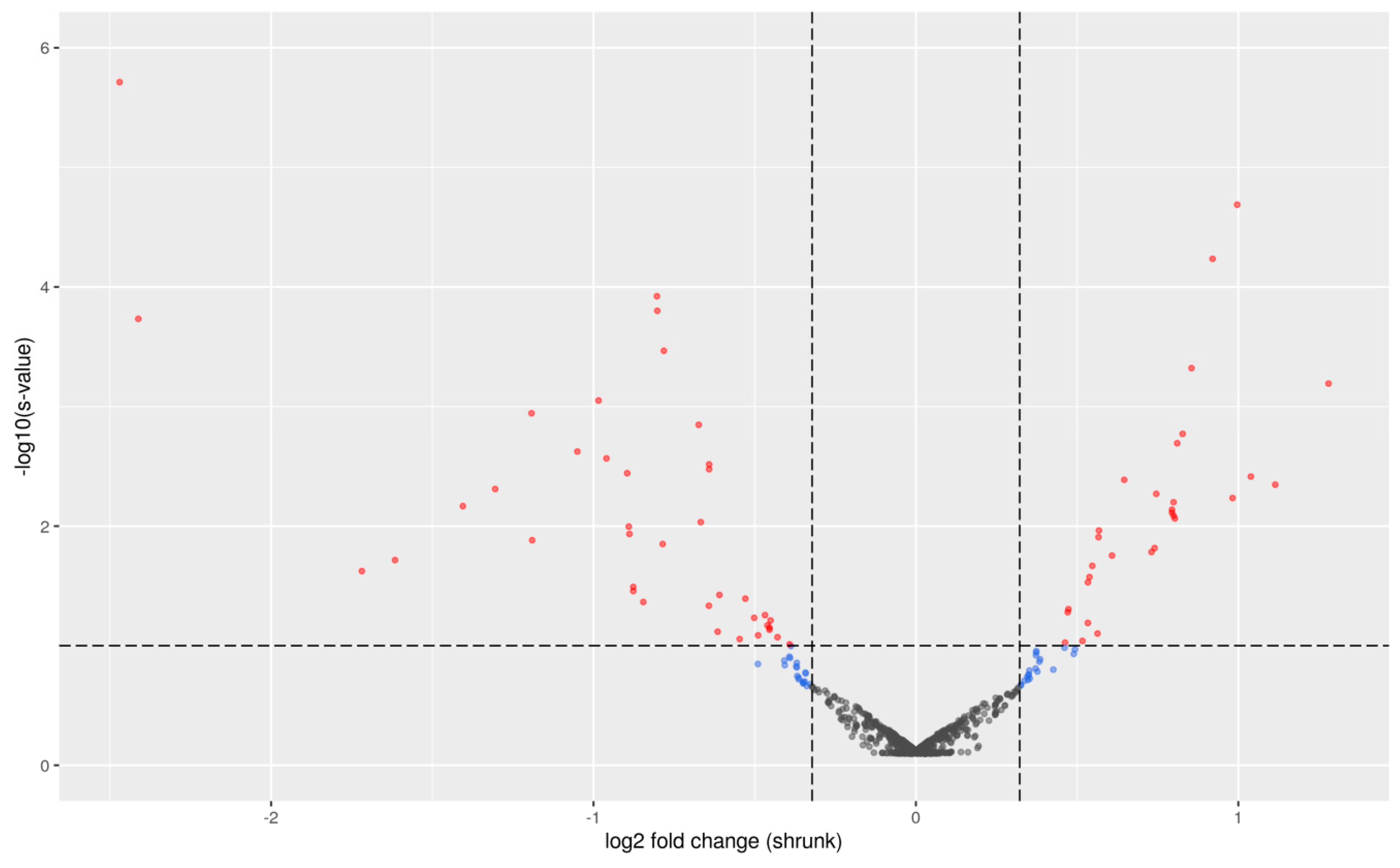
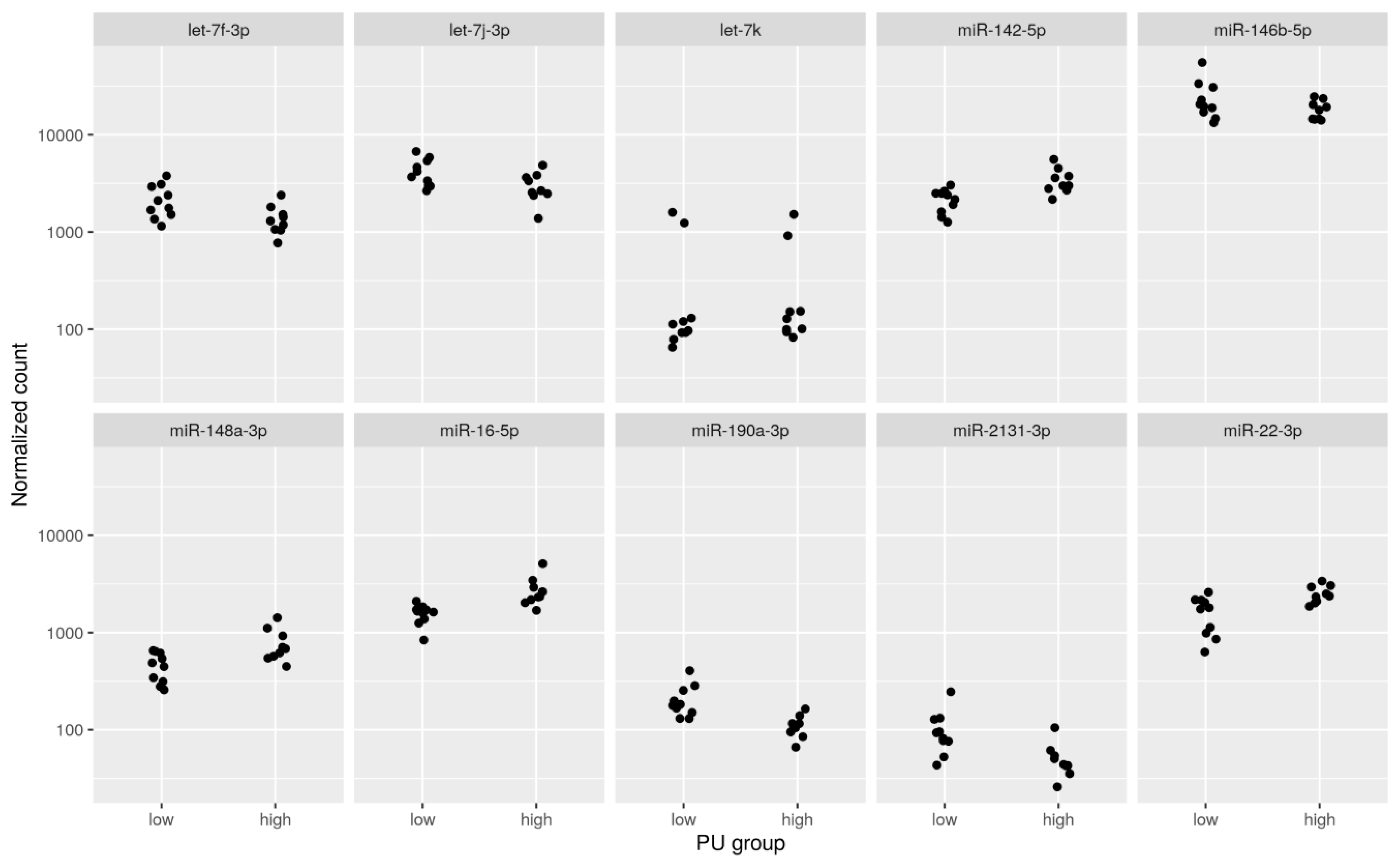
2.1. Differential Expression Analysis between PU Groups
2.2. Correlation between miRNA and mRNA
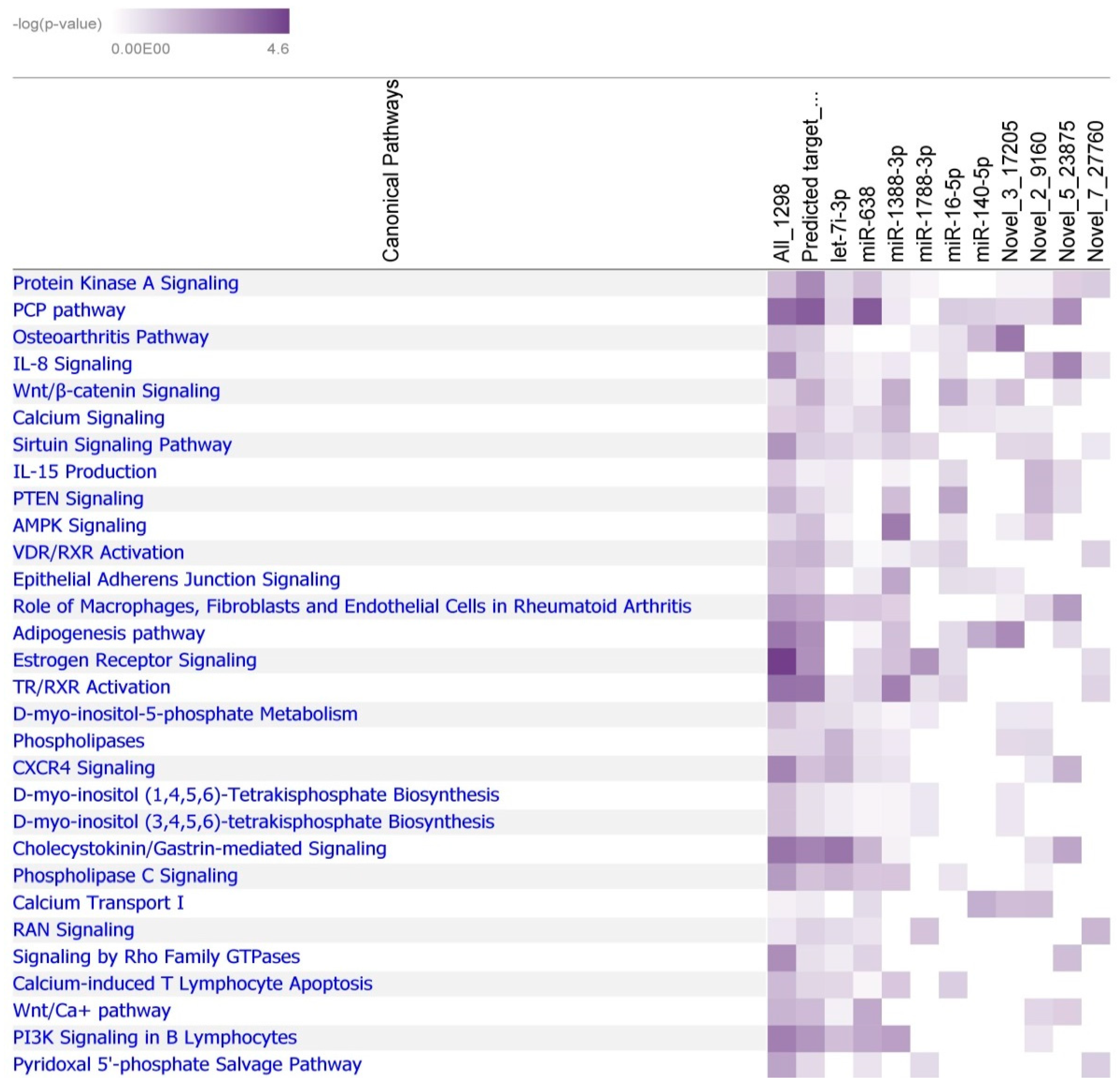
2.3. Prediction of miRNA Targets in Japanese Quail
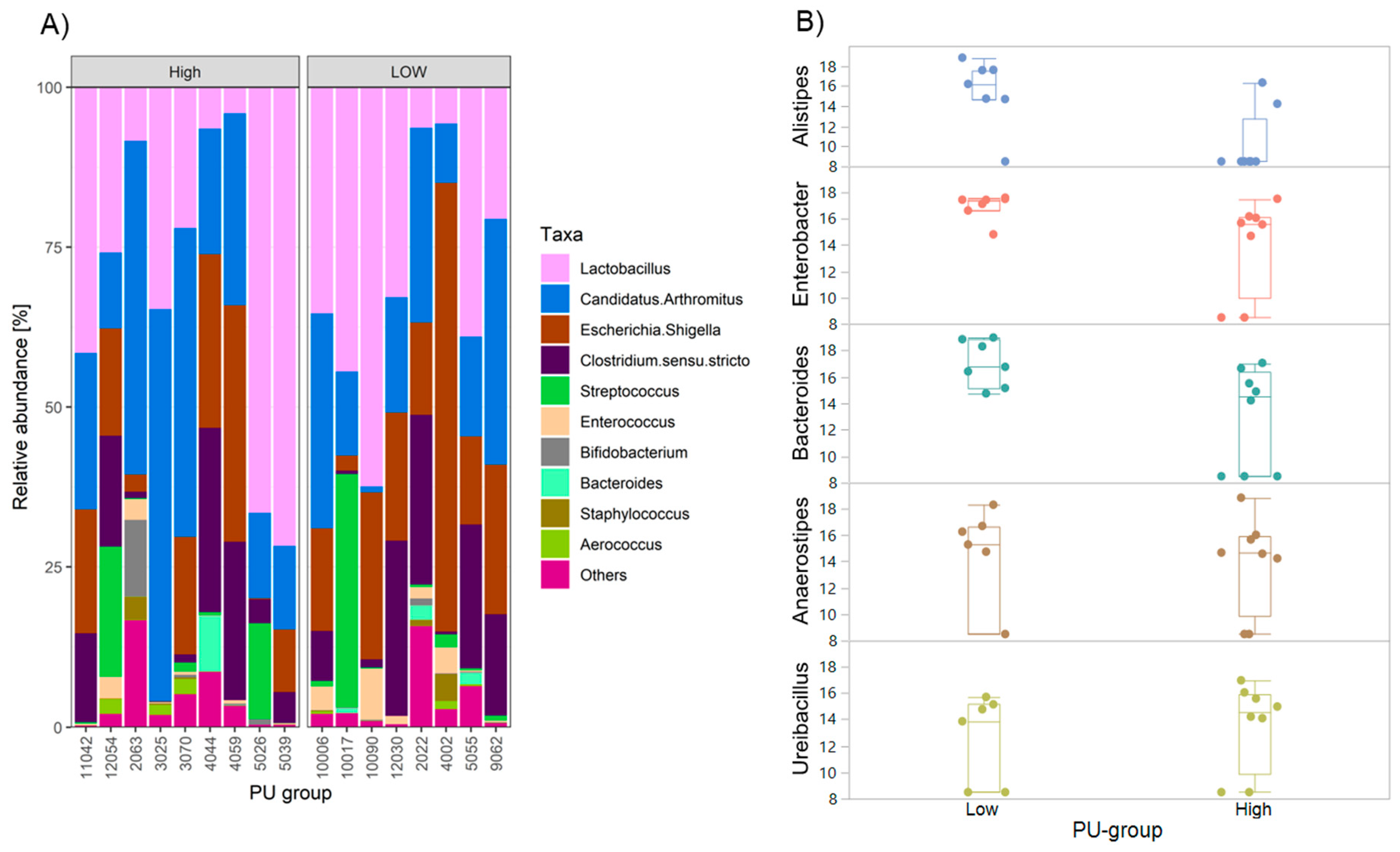
2.4. Differences in Gut Microbiota Composition among PU Group
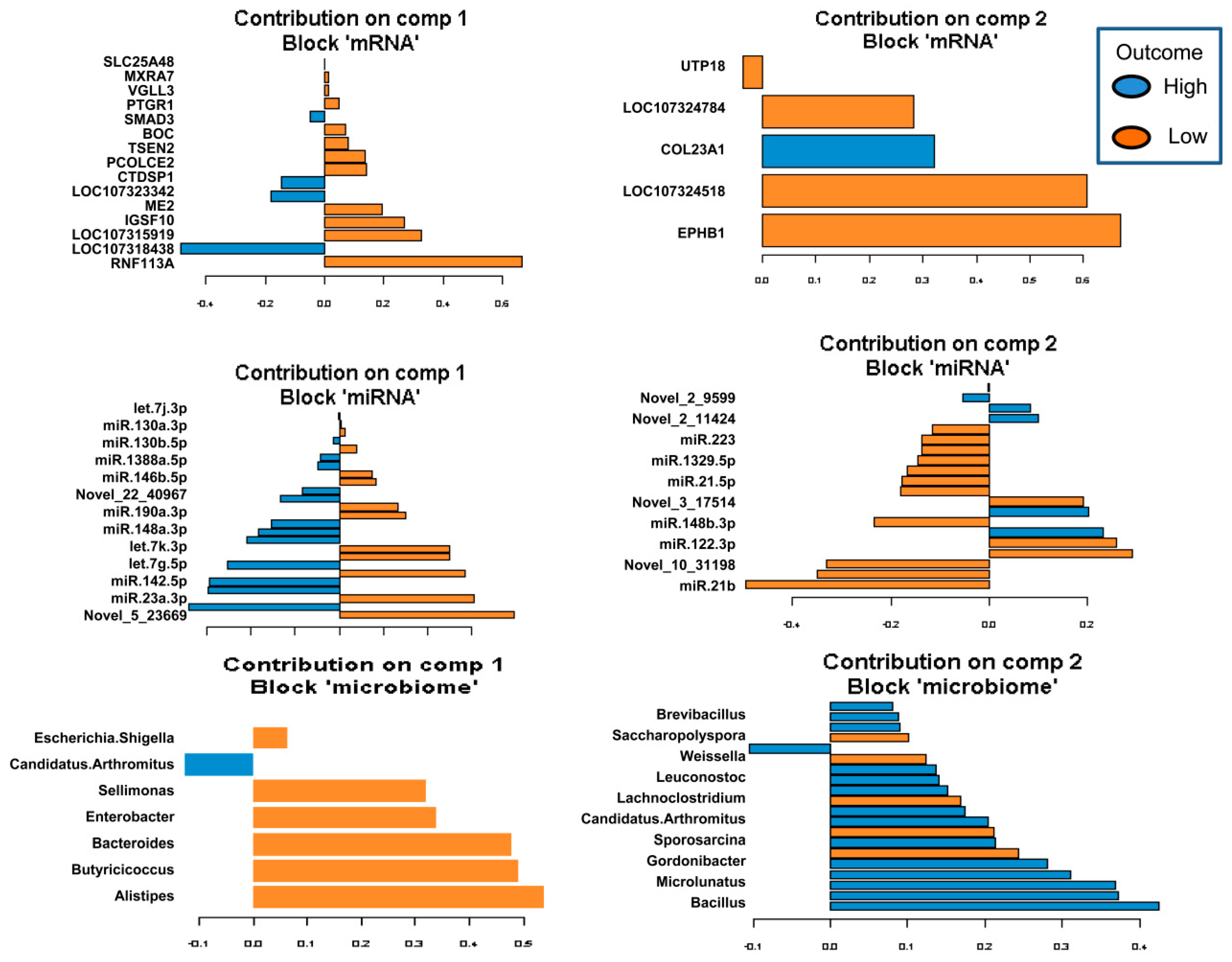
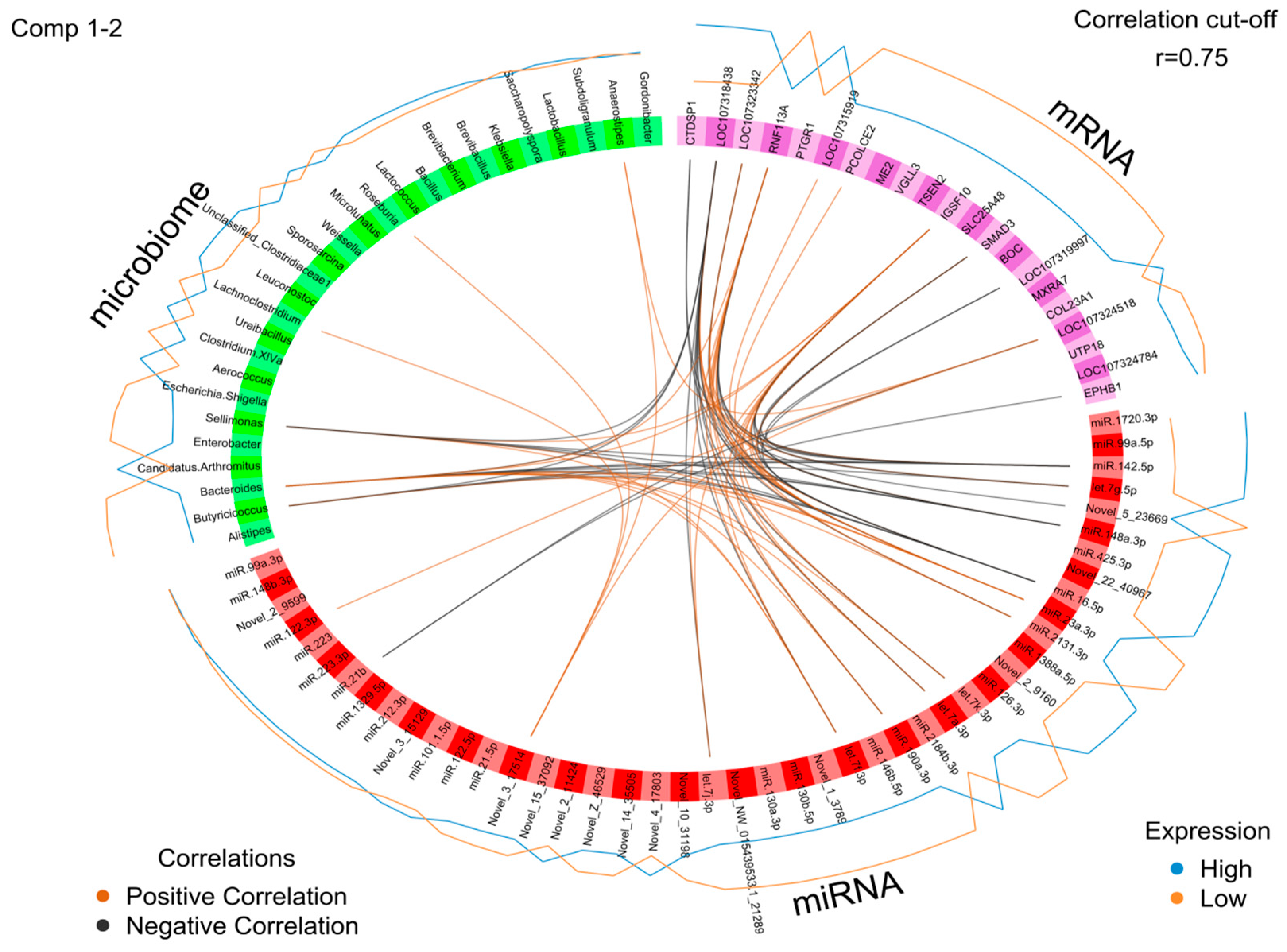
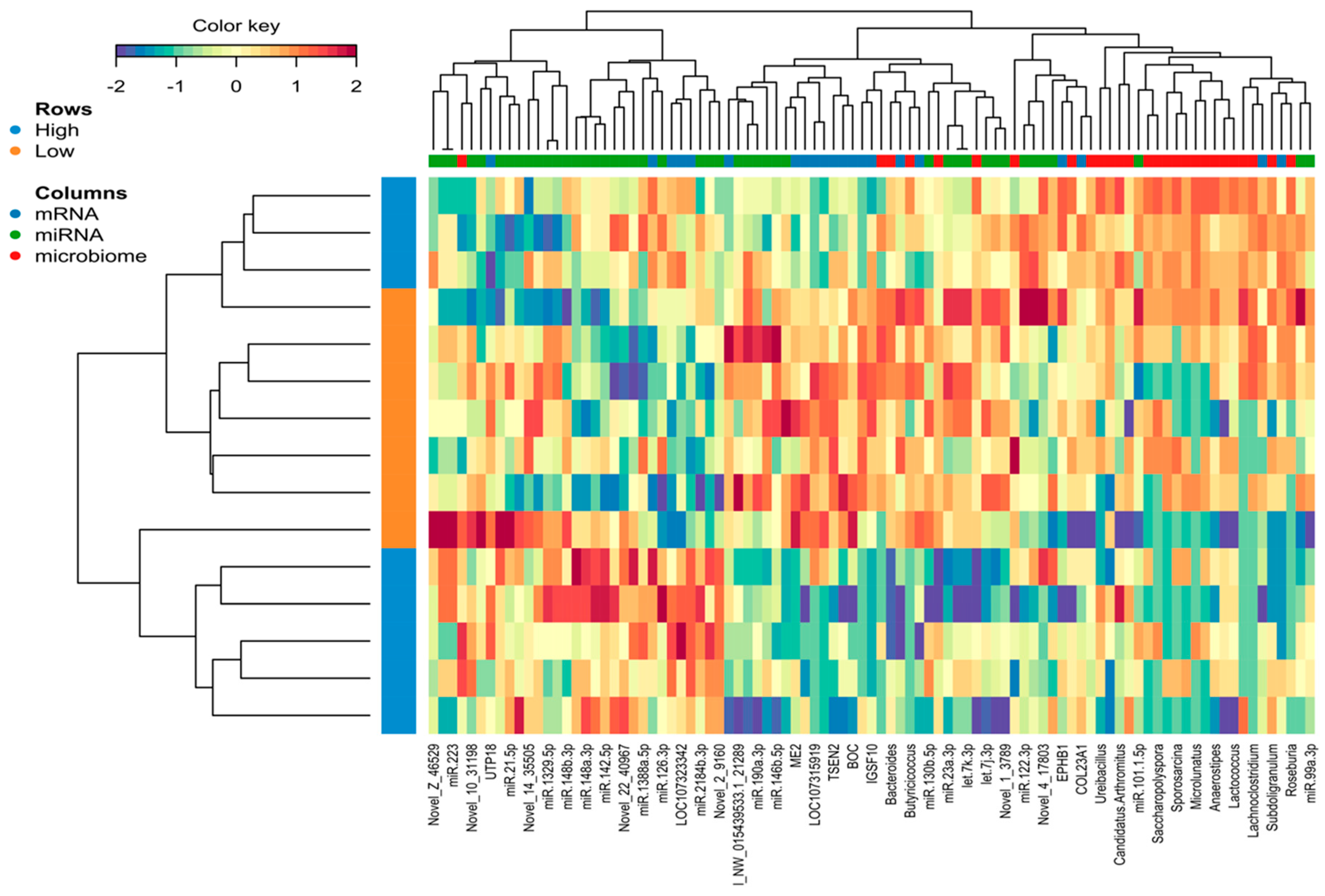
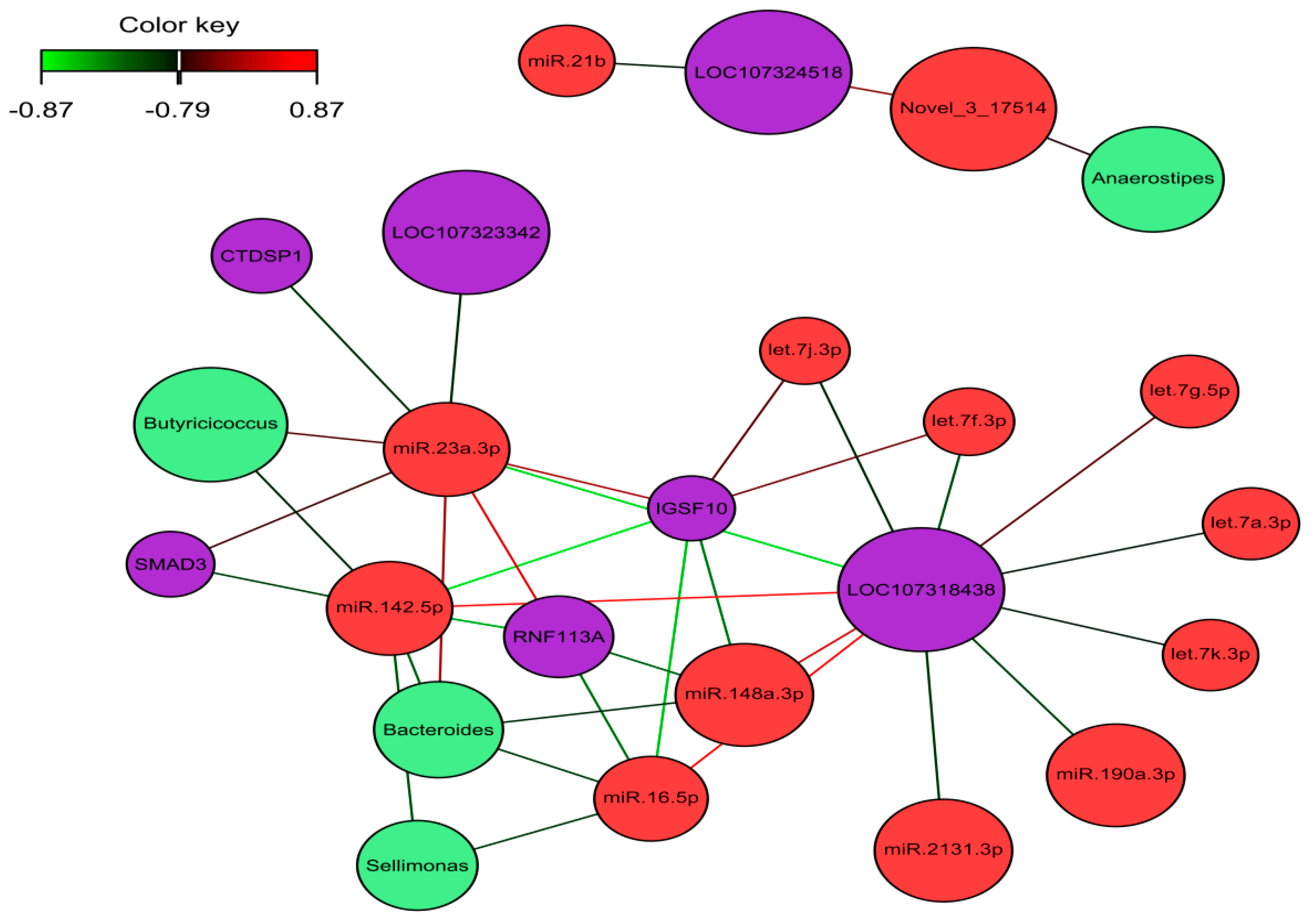
2.5. Identification of the Molecular Drivers of Host–Gut Microbiome-Based PU
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Samples Selection
4.2. RNA Extraction
4.3. Small RNA Library Preparation and Sequencing
4.4. Pre-Processing—Adapter Trimming, Quality Control, and Read Collapsing
4.5. Data Analysis miRNA
4.6. Prediction of miRNA Targets and Correlation between miRNA and mRNA Profiles in Japanese Quail
4.7. Microbiome Data Analysis
4.8. Data Integration of the Microbiota, mRNA, and miRNA
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OTUs | Operational taxonomic units |
| P | Phosphorus |
| PU | Phosphorus utilization |
| OTUs | Operational taxonomic units |
| miRNA | microRNA |
References
- Punna, S.; Roland, D.A. Variation in Phytate Phosphorus Utilization within the Same Broiler Strain. J. Appl. Poult. Res. 1999, 8, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Aggrey, S.E.; Pesti, G.M.; Edwards, H.M.J.; Bakalli, R.I. Genetics of phytate phosphorus bioavailability: Heritability and genetic correlations with growth and feed utilization traits in a randombred chicken population. Poultry Sci. 2003, 82, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Beck, P.; Piepho, H.-P.; Rodehutscord, M.; Bennewitz, J. Inferring relationships between Phosphorus utilization, feed per gain, and bodyweight gain in an F2 cross of Japanese quail using recursive models. Poult. Sci. 2016, 95, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Künzel, S.; Bennewitz, J.; Rodehutscord, M. Genetic parameters for bone ash and phosphorus utilization in an F2 cross of Japanese quail. Poult. Sci. 2019, 98, 4369–4372. [Google Scholar] [CrossRef] [PubMed]
- Reyer, H.; Oster, M.; Wittenburg, D.; Murani, E.; Ponsuksili, S.; Wimmers, K. Genetic Contribution to Variation in Blood Calcium, Phosphorus, and Alkaline Phosphatase Activity in Pigs. Front. Genet. 2019, 10, 590. [Google Scholar] [CrossRef]
- Just, F.; Oster, M.; Büsing, K.; Borgelt, L.; Muráni, E.; Ponsuksili, S.; Wolf, P.; Wimmers, K. Lowered dietary phosphorus affects intestinal and renal gene expression to maintain mineral homeostasis with immunomodulatory implications in weaned piglets. BMC Genom. 2018, 19, 207. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hou, N.; Chen, X.; Li, N.; Zhu, L.; Zhang, Y.; Li, J.; Bian, Z.; Liang, X.; Cai, X.; et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: Evidence of cross-kingdom regulation by microRNA. Cell Res. 2011, 22, 107–126. [Google Scholar] [CrossRef]
- García-Segura, L.; Pérez-Andrade, M.; Miranda-Ríos, J. The Emerging Role of MicroRNAs in the Regulation of Gene Expression by Nutrients. J. Nutr. Nutr. 2013, 6, 16–31. [Google Scholar] [CrossRef]
- Witzig, M.; Camarinha-Silva, A.; Green-Engert, R.; Hoelzle, K.; Zeller, E.; Seifert, J.; Hoelzle, L.E.; Rodehutscord, M. Spatial variation of the gut microbiota in broiler chickens as affected by dietary available phosphorus and assessed by T-RFLP analysis and 454 pyrosequencing. PLoS ONE 2015, 10, e0143442. [Google Scholar]
- Nielsen, T.S.; Bendiks, Z.; Thomsen, B.; Wright, M.E.; Theil, P.; Scherer, B.; Marco, M. High-Amylose Maize, Potato, and Butyrylated Starch Modulate Large Intestinal Fermentation, Microbial Composition, and Oncogenic miRNA Expression in Rats Fed A High-Protein Meat Diet. Int. J. Mol. Sci. 2019, 20, 2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommerfeld, V.; Künzel, S.; Schollenberger, M.; Kühn, I.; Rodehutscord, M. Influence of phytase or myo-inositol supplements on performance and phytate degradation products in the crop, ileum, and blood of broiler chickens. Poult. Sci. 2018, 97, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.-F.; Xiong, Q.; Ge, W.; Zhang, L. The role of MicroRNAs in Osteoclasts and Osteoporosis. RNA Boil. 2014, 11, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.; Tang, J.; He, H.; Cheng, P.; Chen, C. MiR-142-5p promotes bone repair by maintaining osteoblast activity. J. Bone Miner. Metab. 2016, 35, 255–264. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, M.; Zhang, X.; Xu, J.; Hu, G.; Zhao, X.; Cui, P.; Zhang, X. MiR-146a Deletion Protects from Bone Loss in OVX Mice by Suppressing RANKL/OPG and M-CSF in Bone Microenvironment. J. Bone Miner. Res. 2019. [Google Scholar] [CrossRef]
- Ponsuksili, S.; Tesfaye, D.; Schellander, K.; Hoelker, M.; Hadlich, F.; Schwerin, M.; Wimmers, K.; Agrawal, V.; Jaiswal, M.K.; Ilievski, V.; et al. Differential Expression of miRNAs and Their Target mRNAs in Endometria Prior to Maternal Recognition of Pregnancy Associates with Endometrial Receptivity for In Vivo- and In Vitro-Produced Bovine Embryos1. Boil. Reprod. 2014, 91, 135. [Google Scholar] [CrossRef]
- Siengdee, P.; Trakooljul, N.; Murani, E.; Schwerin, M.; Wimmers, K.; Ponsuksili, S. MicroRNAs Regulate Cellular ATP Levels by Targeting Mitochondrial Energy Metabolism Genes during C2C12 Myoblast Differentiation. PLoS ONE 2015, 10, e0127850. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Trakooljul, N.; Hadlich, F.; Muráni, E.; Wimmers, K.; Ponsuksili, S. MicroRNA-mRNA regulatory networking fine-tunes the porcine muscle fiber type, muscular mitochondrial respiratory and metabolic enzyme activities. BMC Genom. 2016, 17, 531. [Google Scholar] [CrossRef]
- Oster, M.; Reyer, H.; Trakooljul, N.; Weber, F.; Xi, L.; Muráni, E.; Ponsuksili, S.; Rodehutscord, M.; Bennewitz, J.; Wimmers, K. Ileal transcriptome profiles of Japanese quail divergent in phosphorus utilization. Int. J. Mol. Sci. 2020, 21, 2762. [Google Scholar] [CrossRef]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Le Cao, K.-A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinform 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Wubuli, A.; Reyer, H.; Muráni, E.; Ponsuksili, S.; Wolf, P.; Oster, M.; Wimmers, K. Tissue-Wide Gene Expression Analysis of Sodium/Phosphate Co-Transporters in Pigs. Int. J. Mol. Sci. 2019, 20, 5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Z.; Zhang, G.; Zhang, J.; Li, J.; Yang, Y. Effect of early dietary energy restriction and phosphorus level on subsequent growth performance, intestinal phosphate transport, and AMPK activity in young broilers. PLoS ONE 2017, 12, e0186828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [Green Version]
- Robling, A.G. The expanding role of Wnt signaling in bone metabolism. Bone 2013, 55, 256. [Google Scholar] [CrossRef] [Green Version]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef]
- Oster, M.; Just, F.; Büsing, K.; Wolf, P.; Polley, C.; Vollmar, B.; Muráni, E.; Ponsuksili, S.; Wimmers, K. Toward improved phosphorus efficiency in monogastrics-interplay of serum, minerals, bone, and immune system after divergent dietary phosphorus supply in swine. Am. J. Physiol. Integr. Comp. Physiol. 2016, 310, R917–R925. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.H.; Ho, J.C.; Li, S.C.; Chen, J.F.; Hsiao, C.C.; Lee, C.H. MiR-146a-5p Expression in Peripheral CD14⁺ Monocytes from Patients with Psoriatic Arthritis Induces Osteoclast Activation, Bone Resorption, and Correlates with Clinical Response. J. Clin. Med. 2019, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Anzola, A.; González, R.; Gámez-Belmonte, R.; Ocón, B.; Aranda, C.J.; Martínez-Moya, P.; López-Posadas, R.; Hernández-Chirlaque, C.; Sánchez de Medina, F.; Martínez-Augustin, O. mR-146a regulates the crosstalk between intestinal epithelial cells, microbial components and inflammatory stimuli. Sci. Rep. 2018, 8, 17350. [Google Scholar] [CrossRef]
- Nakasa, T.; Shibuya, H.; Nagata, Y.; Niimoto, T.; Ochi, M. The inhibitory effect of microRNA-146a expression on bone destruction in collagen-induced arthritis. Arthritis Rheum. 2011, 63, 1582–1590. [Google Scholar] [CrossRef]
- Kim, K.I.; Jeong, S.; Han, N.; Oh, J.M.; Oh, K.H.; Kim, I.W. Identification of differentially expressed miRNAs associated with chronic kidney disease-mineral bone disorder. Front Med. 2017, 11, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Krallis, P.Ν.; Kämpe, A.; Karavitakis, E.M.; Taylan, F.; Mäkitie, O.; Doulgeraki, A. A novel frameshift deletion in PLS3 causing severe primary osteoporosis. J. Hum. Genet. 2018, 63, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Fahiminiya, S.; Majewski, J.; Al-Jallad, H.; Moffatt, P.; Mort, J.; Glorieux, F.H.; Roschger, P.; Klaushofer, K.; Rauch, F. Osteoporosis caused by mutations in PLS3: Clinical and bone tissue characteristics. J. Bone Miner Res. 2014, 29, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, J.; Heilig, J.; Hosseinibarkooie, S.; Ross, B.C.; Mendoza-Ferreira, N.; Nolte, F.; Peters, M.; Hölker, I.; Hupperich, K.; Tschanz, T.; et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum. Mol. Genet. 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Baniwal, S.K.; Khalid, O.; Gabet, Y.; Shah, R.; Purcell, D.J.; Mav, D.; E Kohn-Gabet, A.; Shi, Y.; Coetzee, G.A.; Frenkel, B. Runx2 transcriptome of prostate cancer cells: Insights into invasiveness and bone metastasis. Mol. Cancer 2010, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, A.J.; Leong, D.T.K.; Farquharson, C. PLA 2 and ENPP6 may act in concert to generate phosphocholine from the matrix vesicle membrane during skeletal mineralization. FASEB J. 2017, 32, 20–25. [Google Scholar] [CrossRef]
- Xie, J.; Liu, Y.; Chen, B.; Zhang, G.; Ou, S.; Luo, J.; Peng, X. Ganoderma lucidum polysaccharide improves rat DSS-induced colitis by altering cecal microbiota and gene expression of colonic epithelial cells. Food Nutr. Res. 2019, 63, 63. [Google Scholar] [CrossRef]
- Kelch, S.; Balmayor, E.R.; Seeliger, C.; Vester, H.; Kirschke, J.S.; Van Griensven, M. miRNAs in bone tissue correlate to bone mineral density and circulating miRNAs are gender independent in osteoporotic patients. Sci. Rep. 2017, 7, 15861. [Google Scholar] [CrossRef]
- Manochantr, S.; Marupanthorn, K.; Tantrawatpan, C.; Kheolamai, P.; Tantikanlayaporn, D.; Sanguanjit, P. The Effects of BMP-2, miR-31, miR-106a, and miR-148a on Osteogenic Differentiation of MSCs Derived from Amnion in Comparison with MSCs Derived from the Bone Marrow. Stem Cells Int. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Seeliger, C.; Karpinski, K.; Haug, A.T.; Vester, H.; Schmitt, A.; Bauer, J.S.; Van Griensven, M. Five Freely Circulating miRNAs and Bone Tissue miRNAs Are Associated with Osteoporotic Fractures. J. Bone Miner. Res. 2014, 29, 1718–1728. [Google Scholar] [CrossRef]
- Grieco, G.E.; Cataldo, D.; Ceccarelli, E.; Nigi, L.; Catalano, G.; Brusco, N.; Mancarella, F.; Ventriglia, G.; Fondelli, C.; Guarino, E.; et al. Serum Levels of miR-148a and miR-21-5p Are Increased in Type 1 Diabetic Patients and Correlated with Markers of Bone Strength and Metabolism. Non-Coding RNA 2018, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Talebi, F.; Ghorbani, S.; Chan, W.F.; Boghozian, R.; Masoumi, F.; Ghasemi, S.; Vojgani, M.; Power, C.; Noorbakhsh, F. MicroRNA-142 regulates inflammation and T cell differentiation in an animal model of multiple sclerosis. J. Neuroinflammation 2017, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Al-Quraishy, S.; Delic, D.; Sies, H.; Wunderlich, F.; Abdel-Baki, A.A.; Dkhil, M.A. Differential miRNA expression in the mouse jejunum during garlic treatment of Eimeria papillata infections. Parasitol. Res. 2011, 109, 387–394. [Google Scholar] [CrossRef]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.K.; Shelby, H.; Graham, D.Y.; Streckfus, C.F.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, X.Q.; Liu, X.T.; Guo, R.; Zhang, R.D. Integrative analysis reveals key mRNAs and lncRNAs in monocytes of osteoporotic patients. Math. Biosci. Eng. 2019, 16, 5947–5971. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R. Bone Remodeling and the Microbiome. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.J. Bone Mechanical Function and the Gut Microbiota. Adv. Exp. Med. Biol. 2017. [Google Scholar]
- Hernandez, C.J.; Guss, J.D.; Luna, M.; Goldring, S.R. Links Between the Microbiome and Bone. J. Bone Miner. Res. 2016, 31, 1638–1646. [Google Scholar] [CrossRef]
- Charles, J.F.; Ermann, J.; Aliprantis, A.O. The intestinal microbiome and skeletal fitness: Connecting bugs and bones. Clin. Immunol. 2015, 159, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.R.; Stedtfeld, R.D.; Tiedje, J.M.; Hashsham, S.A. MicroRNAs-Based Inter-Domain Communication between the Host and Members of the Gut Microbiome. Front. Microbiol. 2017, 8, 1896. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Zheng, C.; Li, H. Inhibition of miR-23a-3p promotes osteoblast proliferation and differentiation. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.; Kattamuri, C.; Luedeke, D.M.; Angerman, E.B.; Rankin, S.A.; Stevens, M.L.; Zorn, A.M.; Thompson, T.B. Structure of Neuroblastoma Suppressor of Tumorigenicity 1 (NBL1). J. Boil. Chem. 2015, 290, 4759–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, G; Knockaert, M.; Alarcón, C.; Montalvo, E.; Brivanlou, A.H.; Massagué, J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 2006, 281, 40412–40419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devriese, S.; Eeckhaut, V.; Geirnaert, A.; Hindryckx, P.; Ducatelle, R.; Laukens, D.; Bossche, L.V.D.; Van De Wiele, T.; Van Immerseel, F.; De Vos, M. Reduced mucosa-associated Butyricicoccus activity in patients with ulcerative colitis correlates with aberrant claudin-1 expression. J. Crohn’s Colitis 2016, 11, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Ponsuksili, S.; Trakooljul, N.; Hadlich, F.; Haack, F.; Muráni, E.; Wimmers, K. Genetic architecture and regulatory impact on hepatic microRNA expression linked to immune and metabolic traits. Open Boil. 2017, 7, 170101. [Google Scholar] [CrossRef] [Green Version]
- Friedländer, M.R.; Mackowiak, S.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Ibrahim, J.G.; I Love, M. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinform 2019, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Borda-Molina, D.; Roth, C.; Hemandez-Arriaga, A.; Rissi, D.; Vollmar, S.; Rodehutscord, M.; Bennewitz, J.; Camarinha-Silva, A. P-Fowl: Effects on the ileum microbiota of phosphorus and calcium utilization, bird performance and gender in an F2 cross of Japanese quail. Animals 2020. submitted. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Family | Sex | P-Utilization | Groups | Total Reads Counts (M) | Mapped Reads (M) | Unmapped Reads (M) | % Mapped | % Unmapped |
|---|---|---|---|---|---|---|---|---|---|
| 2063 | 1 | Female | 79.67 | High | 4,643,858 | 3,152,958 | 1,490,900 | 67.90 | 32.11 |
| 4002 | 1 | Female | 50.81 | Low | 3,905,920 | 3,002,397 | 903,523 | 76.87 | 23.13 |
| 5039 | 2 | Female | 76.20 | High | 7,514,630 | 4,940,662 | 2,573,968 | 65.75 | 34.25 |
| 10,006 | 2 | Female | 43.06 | Low | 6,148,717 | 4,457,473 | 1,691,244 | 72.49 | 27.51 |
| 5026 | 3 | Female | 83.43 | High | 6,311,029 | 3,986,110 | 2,324,919 | 63.16 | 36.84 |
| 7023 | 3 | Female | 52.59 | Low | 3,977,952 | 2,849,897 | 1,128,055 | 71.64 | 28.36 |
| 3025 | 4 | Male | 79.16 | High | 4,625,821 | 3,029,840 | 1,595,981 | 65.50 | 34.50 |
| 10,017 | 4 | Male | 39.75 | Low | 4,229,818 | 2,847,346 | 1,382,472 | 67.32 | 32.69 |
| 5055 | 4 | Male | 44.71 | Low | 5,273,807 | 4,082,562 | 1,191,245 | 77.41 | 22.59 |
| 11,042 | 5 | Male | 79.00 | High | 7,237,297 | 4,880,845 | 2,356,452 | 67.44 | 32.56 |
| 3070 | 6 | Male | 86.77 | High | 9,339,712 | 6,706,669 | 2,633,043 | 71.81 | 28.19 |
| 12,030 | 6 | Male | 21.49 | Low | 4,751,504 | 3,588,653 | 1,162,851 | 75.53 | 24.47 |
| 12,054 | 7 | Male | 77.83 | High | 5,903,443 | 3,990,231 | 1,913,212 | 67.60 | 32.41 |
| 6039 | 7 | Male | 27.77 | Low | 3,881,743 | 2,581,642 | 1,300,101 | 66.51 | 33.50 |
| 4059 | 8 | Female | 77.02 | High | 9,276,166 | 6,402,170 | 2,873,996 | 69.02 | 30.99 |
| 2022 | 8 | Female | 45.65 | Low | 5,061,335 | 3,524,773 | 1,536,562 | 69.64 | 30.36 |
| 10,090 | 9 | Female | 48.29 | Low | 4,239,513 | 2,835,088 | 1,404,425 | 66.87 | 33.13 |
| 8017 | 10 | Male | 76.26 | High | 7,515,392 | 5,416,718 | 2,098,674 | 72.08 | 27.93 |
| 6035 | 10 | Male | 24.93 | Low | 5,011,594 | 3,633,621 | 1,377,973 | 72.50 | 27.49 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponsuksili, S.; Reyer, H.; Hadlich, F.; Weber, F.; Trakooljul, N.; Oster, M.; Siengdee, P.; Muráni, E.; Rodehutscord, M.; Camarinha-Silva, A.; et al. Identification of the Key Molecular Drivers of Phosphorus Utilization Based on Host miRNA-mRNA and Gut Microbiome Interactions. Int. J. Mol. Sci. 2020, 21, 2818. https://doi.org/10.3390/ijms21082818
Ponsuksili S, Reyer H, Hadlich F, Weber F, Trakooljul N, Oster M, Siengdee P, Muráni E, Rodehutscord M, Camarinha-Silva A, et al. Identification of the Key Molecular Drivers of Phosphorus Utilization Based on Host miRNA-mRNA and Gut Microbiome Interactions. International Journal of Molecular Sciences. 2020; 21(8):2818. https://doi.org/10.3390/ijms21082818
Chicago/Turabian StylePonsuksili, Siriluck, Henry Reyer, Frieder Hadlich, Frank Weber, Nares Trakooljul, Michael Oster, Puntita Siengdee, Eduard Muráni, Markus Rodehutscord, Amélia Camarinha-Silva, and et al. 2020. "Identification of the Key Molecular Drivers of Phosphorus Utilization Based on Host miRNA-mRNA and Gut Microbiome Interactions" International Journal of Molecular Sciences 21, no. 8: 2818. https://doi.org/10.3390/ijms21082818
APA StylePonsuksili, S., Reyer, H., Hadlich, F., Weber, F., Trakooljul, N., Oster, M., Siengdee, P., Muráni, E., Rodehutscord, M., Camarinha-Silva, A., Bennewitz, J., & Wimmers, K. (2020). Identification of the Key Molecular Drivers of Phosphorus Utilization Based on Host miRNA-mRNA and Gut Microbiome Interactions. International Journal of Molecular Sciences, 21(8), 2818. https://doi.org/10.3390/ijms21082818