Identification and Annotation of Potential Function of Regulatory Antisense Long Non-Coding RNAs Related to Feed Efficiency in Bos taurus Bulls

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Alignment and Mapping of RNA Sequencing Data
2.2. Long Non-Coding RNA Prediction
2.3. Differential Metabolite Abundance
2.4. Set of Prioritized Loci for Co-Expression Network
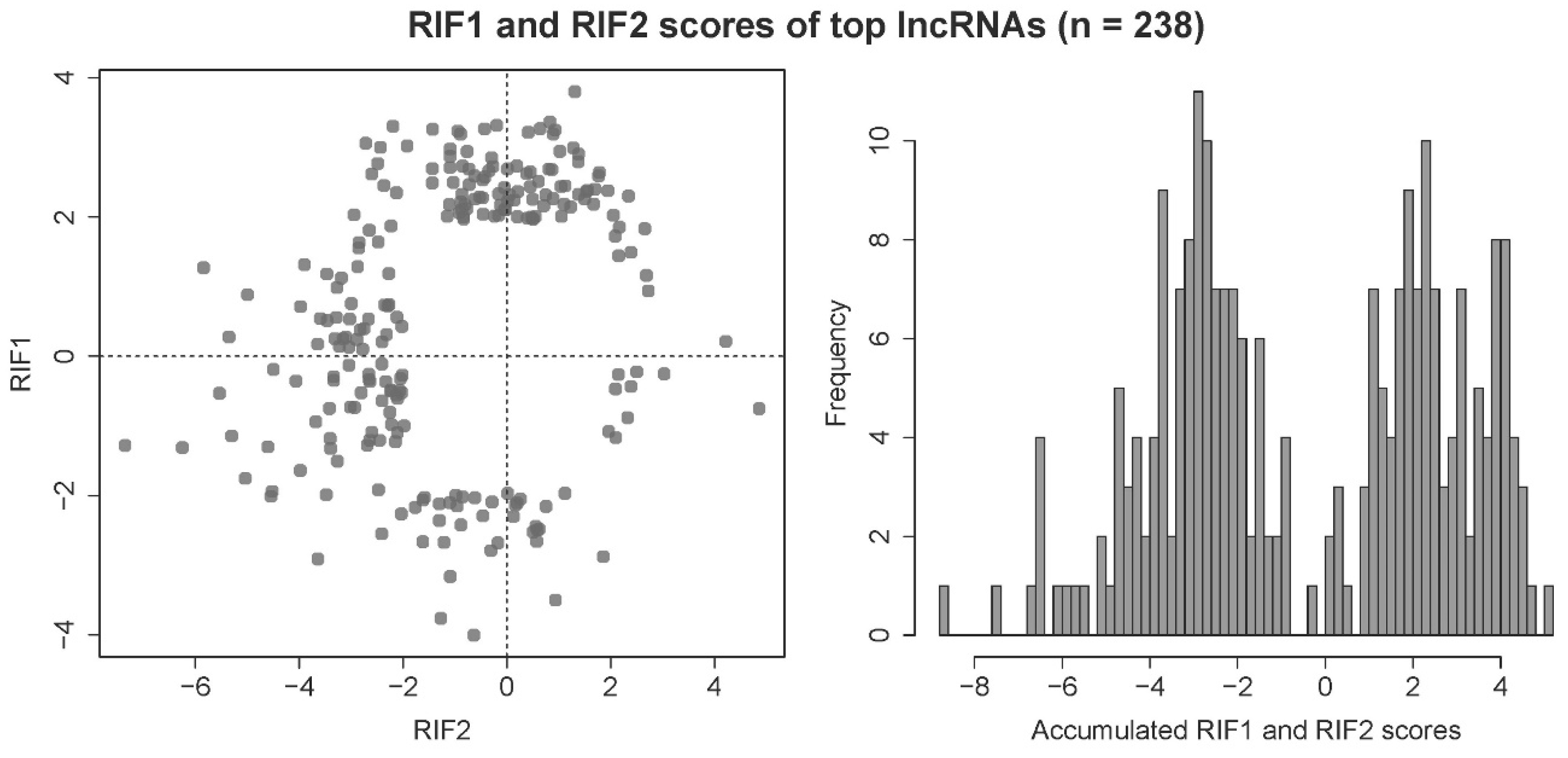
2.5. Regulatory Impact Factor Analysis
2.6. Co-Expression Networks Based on Partial Correlation and Information Theory Approach and Detection of Hub LncRNAs
2.7. Natural Antisense Transcripts
2.8. Characteristics of Key Regulatory Long Non-Coding RNAs in the Co-Expression Network
2.9. Pathway Enrichment Analysis
3. Discussion
3.1. LncRNAs Participating in Fatty Acid β-Oxidation and TCA-Cycle
3.2. LncRNA Linked to Mitochondrial Function and Energy Metabolism
3.3. LncRNA Associated with Immunological Functions
3.4. LncRNAs Putatively Involved in Gluconeogenesis
3.5. LncRNAs as Natural Antisense Transcripts
4. Materials and Methods
4.1. Animals
4.2. Plasma Metabolites
4.3. Sampling, RNA Isolation, Library Preparation, and Sequencing
4.4. Alignment and Assembly
4.5. Long Non-Coding RNA Prediction and Fragment Counting
4.6. Loci Set Prioritization
4.7. Regulatory Impact Factor Analysis
4.8. Partial Correlation and Information Theory
4.9. Correlation of Plasma Metabolites with Key LncRNAs
4.10. Natural Antisense Transcripts
4.11. Selection of Hub Key lncRNAs in Co-Expression Network
4.12. Cis-Action of Hub LncRNAs
4.13. Pathway Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| lncRNA | long non-coding RNA |
| RFI | residual feed intake |
| QTL | quantitative trait locus |
| NAT | natural antisense transcript |
| FAANG | Functional Annotation of Animal Genomes |
| RIF | regulatory impact factor |
| PCIT | partial correlation and information theory |
| nt | nucleotide |
| FC | foldchange |
| PCA | principal component analysis |
| DE | differentially expressed |
| FPKM | fragments per kilobase million |
| IPA | Ingenuity Pathway Analysis |
| SD | standard deviation |
| CF | carcass fat |
| IMF | intramuscular fat content |
| Mb | megabase |
References
- Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, D.A.; Fitzsimons, C.; Waters, S.M.; McGee, M. Invited review: Improving feed efficiency of beef cattle – the current state of the art and future challenges. Animal 2018, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saatchi, M.; Beever, J.E.; Decker, J.E.; Faulkner, D.B.; Freetly, H.C.; Hansen, S.L.; Yampara-Iquise, H.; Johnson, K.A.; Kachman, S.D.; Kerley, M.S.; et al. QTLs associated with dry matter intake, metabolic mid-test weight, growth and feed efficiency have little overlap across 4 beef cattle studies. BMC Genom. 2014, 15, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, P.S.; Cesar, A.S.; do Nascimento, M.L.; Chaves, A.S.; Tizioto, P.C.; Tullio, R.R.; Lanna, D.P.; Rosa, A.N.; Sonstegard, T.S.; Mourao, G.B.; et al. Identification of genomic regions associated with feed efficiency in Nelore cattle. BMC Genet. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.G.; Fitzsimons, C.; McClure, M.C.; McKenna, C.; Conroy, S.; Kenny, D.A.; McGee, M.; Waters, S.M.; Morris, D.W. GWAS and eQTL analysis identifies a SNP associated with both residual feed intake and GFRA2 expression in beef cattle. Sci. Rep. 2018, 8, 14301. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Cao, F.; Wang, S.; Sheng, X.; Ma, J. LncRNAs: The Regulator of Glucose and Lipid Metabolism in Tumor Cells. Front. Oncol. 2019, 9, 1099. [Google Scholar] [CrossRef]
- Muret, K.; Désert, C.; Lagoutte, L.; Boutin, M.; Gondret, F.; Zerjal, T.; Lagarrigue, S. Long noncoding RNAs in lipid metabolism: Literature review and conservation analysis across species. BMC Genom. 2019, 20, 882. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, P.; Yang, W.; Ruan, X.; Kiesewetter, K.; Zhu, J.; Cao, H. Integrative Transcriptome Analyses of Metabolic Responses in Mice Define Pivotal LncRNA Metabolic Regulators. Cell Metab. 2016, 24, 627–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradas-Juni, M.; Hansmeier, N.R.; Link, J.C.; Schmidt, E.; Larsen, B.D.; Klemm, P.; Meola, N.; Topel, H.; Loureiro, R.; Dhaouadi, I.; et al. A MAFG-lncRNA axis links systemic nutrient abundance to hepatic glucose metabolism. Nat. Commun. 2020, 11, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, X.S.; Liu, Q.R.; Wei, L. Genome-wide in silico identification and analysis of cis natural antisense transcripts (cis-NATs) in ten species. Nucleic Acids. Res. 2006, 34, 3465–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef]
- Wenric, S.; ElGuendi, S.; Caberg, J.-H.; Bezzaou, W.; Fasquelle, C.; Charloteaux, B.; Karim, L.; Hennuy, B.; Frères, P.; Collignon, J.; et al. Transcriptome-wide analysis of natural antisense transcripts shows their potential role in breast cancer. Sci. Rep. 2017, 7, 17452. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Hu, Y.; Li, X.; Jin, G.; Chen, X.; Chen, G.; Chen, Y.; Huang, S.; Liao, W.; Liao, Y.; et al. Sirt1 Antisense Long Noncoding RNA Promotes Cardiomyocyte Proliferation by Enhancing the Stability of Sirt1. J. Am. Heart Assoc. 2018, 7, e009700. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-Y.; Wang, J.-H.; Wang, J.-L.; Ma, C.X.; Wang, X.-C.; Liu, F.-S. Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells. BMC Genom. 2015, 16, 676. [Google Scholar] [CrossRef] [Green Version]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinf. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Oliveira, G.B.; Regitano, L.C.A.; Cesar, A.S.M.; Reecy, J.M.; Degaki, K.Y.; Poleti, M.D.; Felicio, A.M.; Koltes, J.E.; Coutinho, L.L. Integrative analysis of microRNAs and mRNAs revealed regulation of composition and metabolism in Nelore cattle. BMC Genom. 2018, 19, 16. [Google Scholar] [CrossRef]
- Deng, L.; Wang, J.; Zhang, J. Predicting Gene Ontology Function of Human MicroRNAs by Integrating Multiple Networks. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Bansal, A.; Singh, T.R.; Chauhan, R.S. A novel miRNA analysis framework to analyze differential biological networks. Sci. Rep. 2017, 7, 14604. [Google Scholar] [CrossRef] [Green Version]
- Weikard, R.; Hadlich, F.; Hammon, H.M.; Frieten, D.; Gerbert, C.; Koch, C.; Dusel, G.; Kuehn, C. Long noncoding RNAs are associated with metabolic and cellular processes in the jejunum mucosa of pre-weaning calves in response to different diets. Oncotarget 2018, 9, 21052–21069. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.C.; Zhu, Y.; Chen, W.B.; Lin, L.W.; Chen, D.H.; Huang, J.R.; Pan, K.; Lin, Y.; Wu, B.T.; Dai, Y.; et al. Integrated analysis of long non-coding RNAs and mRNA expression profiles reveals the potential role of lncRNAs in gastric cancer pathogenesis. Int. J. Oncol. 2014, 45, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhtiarizadeh, M.R.; Salami, S.A. Identification and Expression Analysis of Long Noncoding RNAs in Fat-Tail of Sheep Breeds. G3-Genes Genom. Genet. 2019, 9, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, B.; Li, H.; Liu, M.; Wu, J.; Li, M.; Lei, C.; Huang, B.; Chen, H. Characterization of lncRNA–miRNA–mRNA Network to Reveal Potential Functional ceRNAs in Bovine Skeletal Muscle. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Reverter, A.; Hudson, N.J.; Nagaraj, S.H.; Perez-Enciso, M.; Dalrymple, B.P. Regulatory impact factors: Unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics 2010, 26, 896–904. [Google Scholar] [CrossRef]
- Reverter, A.; Chan, E.K. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [Green Version]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological Network Approach for the Identification of Regulatory Long Non-Coding RNAs Associated With Metabolic Efficiency in Cattle. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25. [Google Scholar] [CrossRef] [Green Version]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, P.A.; Kogelman, L.J.A.; Santana, M.H.A.; Passarelli, D.; Pulz, L.H.; Fantinato-Neto, P.; Silva, P.L.; Leme, P.R.; Strefezzi, R.F.; Coutinho, L.L.; et al. Liver transcriptomic networks reveal main biological processes associated with feed efficiency in beef cattle. BMC Genom. 2015, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, L.D.; Eler, J.P.; Pereira, M.A.; Rosa, A.F.; Alexandre, P.A.; Moncau, C.T.; Salvato, F.; Rosa-Fernandes, L.; Palmisano, G.; Ferraz, J.B.S.; et al. Liver proteomics unravel the metabolic pathways related to Feed Efficiency in beef cattle. Sci. Rep. 2019, 9, 5364. [Google Scholar] [CrossRef] [Green Version]
- Mukiibi, R.; Vinsky, M.; Keogh, K.; Fitzsimmons, C.; Stothard, P.; Waters, S.M.; Li, C. Liver transcriptome profiling of beef steers with divergent growth rate, feed intake, or metabolic body weight phenotypes1. J. Anim. Sci. 2019, 97, 4386–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salleh, S.M.; Mazzoni, G.; Lovendahl, P.; Kadarmideen, H.N. Gene co-expression networks from RNA sequencing of dairy cattle identifies genes and pathways affecting feed efficiency. BMC Bioinf. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Tizioto, P.C.; Coutinho, L.L.; Decker, J.E.; Schnabel, R.D.; Rosa, K.O.; Oliveira, P.S.; Souza, M.M.; Mourão, G.B.; Tullio, R.R.; Chaves, A.S.; et al. Global liver gene expression differences in Nelore steers with divergent residual feed intake phenotypes. BMC Genom. 2015, 16, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Beguec, C.; Wucher, V.; Lagoutte, L.; Cadieul, E.; Botherel, N.; Hedan, B.; De Brito, C.; Guillory, A.S.; Andre, C.; Derrien, T.; et al. Characterisation and functional predictions of canine long non-coding RNAs. Sci. Rep. 2018, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muret, K.; Klopp, C.; Wucher, V.; Esquerré, D.; Legeai, F.; Lecerf, F.; Désert, C.; Boutin, M.; Jehl, F.; Acloque, H.; et al. Long noncoding RNA repertoire in chicken liver and adipose tissue. Genet. Sel. Evol. 2017, 49, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Hu, S.; Tu, T.; Huang, Z.; Tang, Q.; Ma, J.; Wang, X.; Li, X.; Zhou, X.; Shuai, S.; et al. Global Long Noncoding RNA and mRNA Expression Changes between Prenatal and Neonatal Lung Tissue in Pigs. Genes 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011, 147. [Google Scholar] [CrossRef] [Green Version]
- Bogdanos, D.P.; Gao, B.; Gershwin, M.E. Liver immunology. Compr. Physiol. 2013, 3, 567–598. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Ehsani, R.; Drabløs, F. Measures of co-expression for improved function prediction of long non-coding RNAs. BMC Bioinf. 2018, 19, 533. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, P.A.; Naval-Sanchez, M.; Porto-Neto, L.R.; Ferraz, J.B.S.; Reverter, A.; Fukumasu, H. Systems Biology Reveals NR2F6 and TGFB1 as Key Regulators of Feed Efficiency in Beef Cattle. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Canovas, A.; Reverter, A.; DeAtley, K.L.; Ashley, R.L.; Colgrave, M.L.; Fortes, M.R.S.; Islas-Trejo, A.; Lehnert, S.; Porto-Neto, L.; Rincon, G.; et al. Multi-Tissue Omics Analyses Reveal Molecular Regulatory Networks for Puberty in Composite Beef Cattle. PLoS ONE 2014, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Reverter, A.; Cánovas, A.; Venus, B.; Anderson, S.T.; Islas-Trejo, A.; Dias, M.M.; Crawford, N.F.; Lehnert, S.A.; Medrano, J.F.; et al. STAT6, PBX2, and PBRM1 Emerge as Predicted Regulators of 452 Differentially Expressed Genes Associated With Puberty in Brahman Heifers. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonso, J.; Fortes, M.R.S.; Reverter, A.; da Silva Diniz, W.J.; Cesar, A.S.M.; de Lima, A.O.; Petrini, J.; de Souza, M.M.; Coutinho, L.L.; Mourão, G.B.; et al. Genetic regulators of mineral amount in Nelore cattle muscle predicted by a new co-expression and regulatory impact factor approach. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cesar, A.S.M.; Regitano, L.C.A.; Koltes, J.E.; Fritz-Waters, E.R.; Lanna, D.P.D.; Gasparin, G.; Mourao, G.B.; Oliveira, P.S.N.; Reecy, J.M.; Coutinho, L.L. Putative Regulatory Factors Associated with Intramuscular Fat Content. PLoS ONE 2015, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, P.S.N.; Coutinho, L.L.; Cesar, A.S.M.; Diniz, W.J.d.S.; de Souza, M.M.; Andrade, B.G.; Koltes, J.E.; Mourão, G.B.; Zerlotini, A.; Reecy, J.M.; et al. Co-Expression Networks Reveal Potential Regulatory Roles of miRNAs in Fatty Acid Composition of Nelore Cattle. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Adam, A.C.; Lie, K.K.; Moren, M.; Skjaerven, K.H. High dietary arachidonic acid levels induce changes in complex lipids and immune-related eicosanoids and increase levels of oxidised metabolites in zebrafish (Danio rerio). Br. J. Nutr. 2017, 117, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kadarmideen, H.N. Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers. Metabolites 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Gondret, F.; Vincent, A.; Houée-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H.; Louveau, I. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, Z.; Lamont, S.J.; Lee, W.R.; Abasht, B. RNA-Seq Analysis of Abdominal Fat Reveals Differences between Modern Commercial Broiler Chickens with High and Low Feed Efficiencies. PLoS ONE 2015, 10, e0135810. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator–activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigors, S.; O’Doherty, J.V.; Bryan, K.; Sweeney, T. A comparative analysis of the transcriptome profiles of liver and muscle tissue in pigs divergent for feed efficiency. BMC Genom. 2019, 20, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haut, S.; Brivet, M.; Touati, G.; Rustin, P.; Lebon, S.; Garcia-Cazorla, A.; Saudubray, J.M.; Boutron, A.; Legrand, A.; Slama, A. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Hum. Genet. 2003, 113, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Holland, B.P.; Mills, R.L.; Maxwell, C.L.; Farney, J.K.; Terrill, S.J.; Step, D.L.; Richards, C.J.; Burciaga Robles, L.O.; Krehbiel, C.R. Growth and Development Symposium: Impacts of inflammation on cattle growth and carcass merit. J. Anim. Sci. 2012, 90, 1438–1451. [Google Scholar] [CrossRef]
- Fassah, D.M.; Jeong, J.Y.; Baik, M. Hepatic transcriptional changes in critical genes for gluconeogenesis following castration of bulls. Asian Austral. J. Anim. Sci. 2018, 31, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.; Kristensen, N.B. Effect of abomasal glucose infusion on splanchnic and whole-body glucose metabolism in periparturient dairy cows. J. Dairy Sci. 2009, 92, 1071–1083. [Google Scholar] [CrossRef]
- Tanaka, A.; Urabe, S.; Takeguchi, A.; Mizutani, H.; Sako, T.; Imai, S.; Yoshimura, I.; Kimura, N.; Arai, T. Comparison of activities of enzymes related to energy metabolism in peripheral leukocytes and livers between Holstein dairy cows and ICR mice. Vet. Res. Commun. 2006, 30, 29–38. [Google Scholar] [CrossRef]
- Latgé, G.; Poulet, C.; Bours, V.; Josse, C.; Jerusalem, G. Natural Antisense Transcripts: Molecular Mechanisms and Implications in Breast Cancers. Int. J. Mol. Sci. 2018, 19, 123. [Google Scholar] [CrossRef] [Green Version]
- Napoli, S.; Piccinelli, V.; Mapelli, S.N.; Pisignano, G.; Catapano, C.V. Natural antisense transcripts drive a regulatory cascade controlling c-MYC transcription. RNA Biol. 2017, 14, 1742–1755. [Google Scholar] [CrossRef]
- Rosikiewicz, W.; Makalowska, I. Biological functions of natural antisense transcripts. Acta biochimica Polonica 2016, 63, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Kühn, C.; Bellmann, O.; Voigt, J.; Wegner, J.; Guiard, V.; Ender, K. An experimental approach for studying the genetic and physiological background of nutrient transformation in cattle with respect to nutrient secretion and accretion type. Arch. Anim. Breed 2002, 45, 317–330. [Google Scholar] [CrossRef]
- Eberlein, A.; Takasuga, A.; Setoguchi, K.; Pfuhl, R.; Flisikowski, K.; Fries, R.; Klopp, N.; Furbass, R.; Weikard, R.; Kuhn, C. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics 2009, 183, 951–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmann, P.; Nuernberg, K.; Kuehn, C.; Weikard, R. Association of an ACSL1 gene variant with polyunsaturated fatty acids in bovine skeletal muscle. BMC Genet. 2011, 12, 96. [Google Scholar] [CrossRef] [Green Version]
- Archer, J.A.; Arthur, P.F.; Herd, R.M.; Parnell, P.F.; Pitchford, W.S. Optimum postweaning test for measurement of growth rate, feed intake, and feed efficiency in British breed cattle. J. Anim. Sci. 1997, 75, 2024–2032. [Google Scholar] [CrossRef]
- Mock, A.; Warta, R.; Dettling, S.; Brors, B.; Jäger, D.; Herold-Mende, C. MetaboDiff: An R package for differential metabolomic analysis. Bioinformatics 2018, 34, 3417–3418. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Royal. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Weikard, R.; Goldammer, T.; Brunner, R.M.; Kuehn, C. Tissue-specific mRNA expression patterns reveal a coordinated metabolic response associated with genetic selection for milk production in cows. Physiol. Genom. 2012, 44, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Andrew, S. FastQC: A quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 28 March 2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Robinson, A. Quality Trim version 1.6.0. Available online: https://bitbucket.org/arobinson/qualitytrim/downloads/ (accessed on 29 March 2018).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Na. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankish, A.; Vullo, A.; Zadissa, A.; Yates, A.; Thormann, A.; Parker, A.; Gall, A.; Moore, B.; Walts, B.; Aken, B.L.; et al. Ensembl 2018. Nucleic Acids Res. 2017, 46, 754–761. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Harrell, F.E., Jr. Hmisc: Harrell Miscellaneous. R Package Version 2019. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Depth [Read Pairs] | Alignment to ARS-UCD.1.2 (%) | Mapping to Project-Specific Annotation (%) | |
|---|---|---|---|
| Mean | 49,831,770 | 98.72 | 85.98 |
| SD | 5,588,004 | 0.26 | 1.40 |
| lncRNA | Position | Structure | Expression (FPKM 3) | Differential Expression Analysis | ||||||||
| Locus ID | BTA 1 | Start bp 2 | End bp | Strand | Number Exons | Exonic Length | Mean | Mean High Efficiency Group | Mean Low Efficiency Group | Log2FC 4 | p-Value | Adjusted p-Value (BH 5) |
| MSTRG.4390 | 14 | 518,688 | 534,106 | - | 2 | 20,919 | 2.586 | 2.672 | 2.507 | 0.0661 | 0.501 | 0.796 |
| MSTRG.4802 | 14 | 67,986,656 | 67,991,285 | - | 5 | 806 | 1.009 | 0.798 | 1.205 | -0.6310 | 0.004 | 0.091 |
| MSTRG.5042 | 15 | 27,503,347 | 27,512,980 | + | 7 | 3,002 | 0.843 | 1.044 | 0.658 | 0.6330 | 0.043 | 0.287 |
| MSTRG.7472 | 18 | 39,037,005 | 39,043,726 | + | 7 | 1,920 | 11.200 | 11.016 | 11.370 | -0.1053 | 0.886 | 0.966 |
| lncRNA | FEELnc Analysis | cis Action | ||||||||||
| Locus ID | Best Potential Partner Gene | Direction | Type | Distance | Subtype Location | Interaction Partner Gene | PCIT (r) 7 | Direction | ||||
| MSTRG.4390 | ENSBTAG00000046026 | AS 6 | genic | overlapping | exonic | no cis interaction with a minimal correlation of r = 0.65 | ||||||
| MSTRG.4802 | ENSBTAG00000001521 (UQCRB) | AS | genic | nested | exonic | ENSBTAG00000001521 (UQCRB) MSTRG.4780 ENSBTAG00000032432 MSTRG.4798 | 0.690.670.670.67 | antisense sensesense sense | ||||
| MSTRG.5042 | ENSBTAG00000002258 (APOA1) | AS | genic | containing | exonic | ENSBTAG00000002258 (APOA1) | 0.98 | antisense | ||||
| MSTRG.7472 | ENSBTAG00000006354 (HP) | AS | genic | containing | exonic | ENSBTAG00000006354 (HP) | 0.97 | antisense | ||||
| Lnc RNA | Ingenuity Canonical Pathways | −log10(p) | p-Value | Ratio | z-Score | Molecules |
|---|---|---|---|---|---|---|
| MSTRG.4390 | Fatty Acid β-oxidation I | 5.56 | 2.75 × 10−6 | 8.89 × 10−2 | 1.00 | ACADM, ACSL1, ECHS1, HADHB |
| Palmitate Biosynthesis I (Animals) | 3.52 | 3.02 × 10−4 | 1.67 × 10−1 | NaN | lauric acid, palmitic acid | |
| Stearate Biosynthesis I (Animals) | 3.52 | 3.02 × 10−4 | 5.00 × 10−2 | NaN | ACSL1, palmitic acid, stearic acid | |
| Ketolysis | 3.11 | 7.76 × 10−4 | 1.05 × 10−1 | NaN | HADHB, succinic acid | |
| γ-linolenate Biosynthesis II (Animals) | 2.91 | 1.23 × 10−3 | 8.33 × 10−2 | NaN | ACSL1, linoleic acid | |
| MSTRG.4802 | Oxidative Phosphorylation | 7.00 | 1.00 × 10−7 | 4.2 × 10−2 | −2.236 | ATP5MF, ATP5PD, COX5A, NDUFB10, UQCRB |
| Mitochondrial Dysfunction | 6.02 | 9.55 × 10−7 | 2.66 × 10−2 | NaN | ATP5MF, ATP5PD, COX5A, NDUFB10, UQCRB | |
| Spermine Biosynthesis | 2.16 | 6.92 × 10−3 | 1.43 × 10-1 | NaN | SMS | |
| Sirtuin Signaling Pathway | 1.40 | 3.98 × 10−2 | 6.17 × 10−3 | NaN | ATG3, NDUFB10 | |
| TNFR1 Signaling | 1.32 | 4.79 × 10−2 | 2.00 × 10−2 | NaN | MADD | |
| MSTRG.5042 | TCA Cycle II (Eukaryotic) | 3.48 | 3.31 × 10−4 | 7.14 × 10−2 | NaN | fumaric acid, L-malic acid, succinic acid |
| Palmitate Biosynthesis I (Animals) | 3.19 | 6.46 × 10−4 | 1.67 × 10−1 | NaN | lauric acid, palmitic acid | |
| Glycerol Degradation I | 3.12 | 7.59 × 10−4 | 1.54 × 10−1 | NaN | GK, glycerol | |
| Stearate Biosynthesis I (Animals) | 3.03 | 9.33 × 10−4 | 5.00 × 10−2 | NaN | ACSL1, palmitic acid, stearic acid | |
| γ-linolenate Biosynthesis II (Animals) | 2.58 | 2.63 × 10−3 | 8.33 × 10−2 | NaN | ACSL1, linoleic acid | |
| MSTRG.7472 | Acute Phase Response Signaling | 1.12 x 101 | 6.31 × 10−12 | 5.52 × 10−2 | −0.378 | C5, FGG, HP, HPX, HRG, LBP, OSMR, SAA2, SOCS3, STAT3 |
| Unfolded protein response | 6.82 | 1.51 × 10−7 | 8.93 × 10−2 | 0.447 | CANX, DNAJC3, P4HB, PDIA6, XBP1 | |
| Role of JAK family kinases in IL-6-type Cytokine Signaling | 4.64 | 2.29 × 10−5 | 1.20 × 10−1 | NaN | OSMR, SOCS3, STAT3 | |
| Role of JAK2 in Hormone-like Cytokine Signaling | 4.24 | 5.75 × 10−5 | 8.82 × 10−2 | NaN | GHR, SOCS3, STAT3 | |
| Role of Tissue Factor in Cancer | 3.85 | 1.41 × 10−4 | 3.36 × 10−2 | NaN | CFL1, FGG, P4HB, PDIA6 |
| Lnc RNA | Upstream Regulator | Activation z-Score | p-Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|
| MSTRG.4390 | PML | −2.433 | 1.22 × 10−6 | ACADM, APOA1, HADHB, myristic acid, palmitic acid, stearic acid |
| TP53 | 0.113 | 3.21 × 10−2 | ACADM, ACSL1, APOA1, HADHB, IDH1, INHBA, NDRG2, PCK1 | |
| SIRT1 | 0.317 | 8.98 × 10−3 | ACADM, glycerol, MAT2A, PCK1 | |
| MYC | 0.577 | 2.51 × 10−2 | IDH1, INHBA, MAT2A, NDRG2, PCK1, SHMT2 | |
| SREBF1 | 0.652 | 1.69 × 10−3 | ACSL1, ARF4, IDH1, PCK1 | |
| HNF4A | 1.181 | 8.09 × 10−3 | ACSL1, APOA1, HADHB, HSDL2, INHBA, MAT2A, MPP1, PCK1, RAB30, TRIP11 | |
| PPARGC1A | 1.729 | 7.33 × 10−5 | ACADM, INHBA, myristic acid, palmitic acid, PCK1, stearic acid | |
| PPARGC1B | 2.177 | 4.51 × 10−7 | ACADM, myristic acid, palmitic acid, PCK1, stearic acid | |
| MSTRG.4802 | PPARGC1B | 3.03 × 10−3 | ATP5MF, COX5A | |
| ARID5B | 4.15 × 10−3 | UQCRB | ||
| Esrra | 5.68 × 10−3 | ATP5MF, COX5A | ||
| PPARGC1A | 6.22 × 10−3 | ATP5MF, ATP5PD, COX5A | ||
| HNF1A | 1.44 × 10−2 | AP3M1, ATG3, CLTRN | ||
| KMT2D | 1.85 × 10−2 | FBXL21P, PTGR2 | ||
| SUB1 | 2.97 × 10−2 | NDUFB10 | ||
| HTT | 4.36 × 10−2 | AGRN, ATP5MF, UQCRB | ||
| MSTRG.5042 | PML | −2.000 | 1.89 × 10−3 | APOA1, myristic acid, palmitic acid, stearic acid |
| SREBF1 | 0.652 | 7.56 × 10−3 | ACSL1, ARF4, IDH1, PCK1 | |
| TCF7L2 | 0.728 | 2.99 × 10−3 | ACSL1, ADIPOR2, FBP1, IDH1, PCK1 | |
| HNF4A | 1.505 | 1.03 × 10−2 | ACSL1, APOA1, ASGR2, FBP1, HSDL2, INHBA, MAT2A, PABPN1, PCK1, RAB30, RTCB, SOAT2, TRIP11 | |
| PPARGC1A | 1.673 | 7.26 × 10−4 | GK, INHBA, myristic acid, palmitic acid, PCK1, stearic acid | |
| SP1 | 1.934 | 2.66 × 10−2 | ACSL1, APOA1, MAT2A, PCK1, THRB | |
| PPARGC1B | 2.000 | 8.73 × 10−5 | myristic acid, palmitic acid, PCK1, stearic acid | |
| MSTRG.7472 | STAT3 | −0.877 | 6.51 × 10−5 | C5, FGG, HP, LBP, PDIA4, SOCS3, STAT3, XBP1 |
| TP53 | −0.640 | 3.11 × 10−2 | CD44, HDLBP, NARS1, P4HB, PDIA6, STAT3, TMSB10/TMSB4X, UGDH, XBP1 | |
| ATF4 | −0.152 | 3.90 × 10−5 | CANX, NARS1, OSMR, SLC39A14, STAT3 | |
| CEBPB | −0.133 | 5.64 × 10−5 | HP, HPX, LBP, SAA2, SOCS3, STAT3, XBP1 | |
| NFE2L2 | 0.000 | 6.00 × 10−8 | C5, DNAJC3, GHR, NARS1, PDIA4, PDIA6, SOCS3, TMED2, UGDH, XBP1 | |
| XBP1 | 0.262 | 1.16 × 10−6 | DNAJC3, FKBP2, P4HB, PDIA4, PDIA6, SEC61G, XBP1 | |
| ATF6 | 0.762 | 1.50 × 10−5 | DNAJC3, PDIA4, SLC39A14, XBP1 | |
| TCF3 | 1.000 | 6.56 × 10−8 | AZGP1, EPRS1, GPLD1, NUF2, PDIA4, PDIA6, RASSF4, SOCS3, XBP1 | |
| TCF4 | 1.000 | 3.11 × 10−4 | NUF2, PDIA4, PDIA6, SOCS3, STAT3, XBP1 | |
| HNF1A | 1.114 | 1.77 × 10−6 | C5, FGL1, HOPX, HPX, LBP, NUF2, SOCS3, TARS1, XBP1 | |
| PRDM1 | 1.176 | 1.91 × 10−3 | CD44, FGG, TRIB1, XBP1 | |
| HIF1A | 1.932 | 3.21 × 10−3 | CD44, GHR, HP, SOCS3, STAT3 |
| Group | Number of Animals. | CF (%) | IMF (%) | RFI in MJ ME/day | |||
|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | ||
| high efficiency | 13 | 14.39 | 2.86 | 2.77 | 0.95 | −20.91 | 4.47 |
| low efficiency | 13 | 20.28 | 4.06 | 4.59 | 1.71 | 20.48 | 4.40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Identification and Annotation of Potential Function of Regulatory Antisense Long Non-Coding RNAs Related to Feed Efficiency in Bos taurus Bulls. Int. J. Mol. Sci. 2020, 21, 3292. https://doi.org/10.3390/ijms21093292
Nolte W, Weikard R, Brunner RM, Albrecht E, Hammon HM, Reverter A, Kühn C. Identification and Annotation of Potential Function of Regulatory Antisense Long Non-Coding RNAs Related to Feed Efficiency in Bos taurus Bulls. International Journal of Molecular Sciences. 2020; 21(9):3292. https://doi.org/10.3390/ijms21093292
Chicago/Turabian StyleNolte, Wietje, Rosemarie Weikard, Ronald M. Brunner, Elke Albrecht, Harald M. Hammon, Antonio Reverter, and Christa Kühn. 2020. "Identification and Annotation of Potential Function of Regulatory Antisense Long Non-Coding RNAs Related to Feed Efficiency in Bos taurus Bulls" International Journal of Molecular Sciences 21, no. 9: 3292. https://doi.org/10.3390/ijms21093292
APA StyleNolte, W., Weikard, R., Brunner, R. M., Albrecht, E., Hammon, H. M., Reverter, A., & Kühn, C. (2020). Identification and Annotation of Potential Function of Regulatory Antisense Long Non-Coding RNAs Related to Feed Efficiency in Bos taurus Bulls. International Journal of Molecular Sciences, 21(9), 3292. https://doi.org/10.3390/ijms21093292