Deletion of SOCS2 Reduces Post-Colitis Fibrosis via Alteration of the TGFβ Pathway


Abstract
:1. Introduction
2. Results
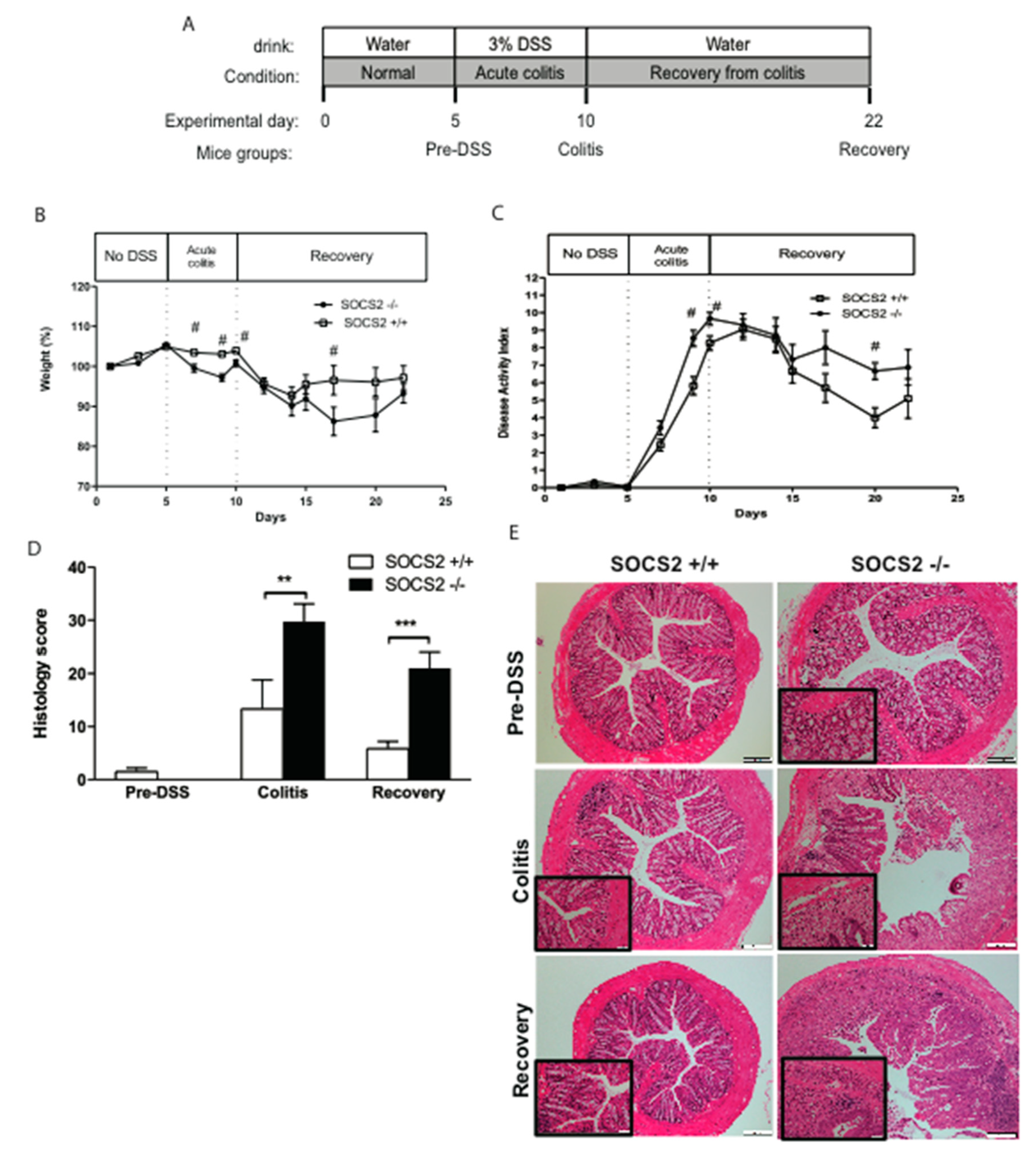
2.1. SOCS2 Deletion Aggravates Colitis Severity in SOCS2−/− Mice
2.2. Higher Recovery Rate in SOCS2−/− Mice Despite an Increased Inflammatory Process
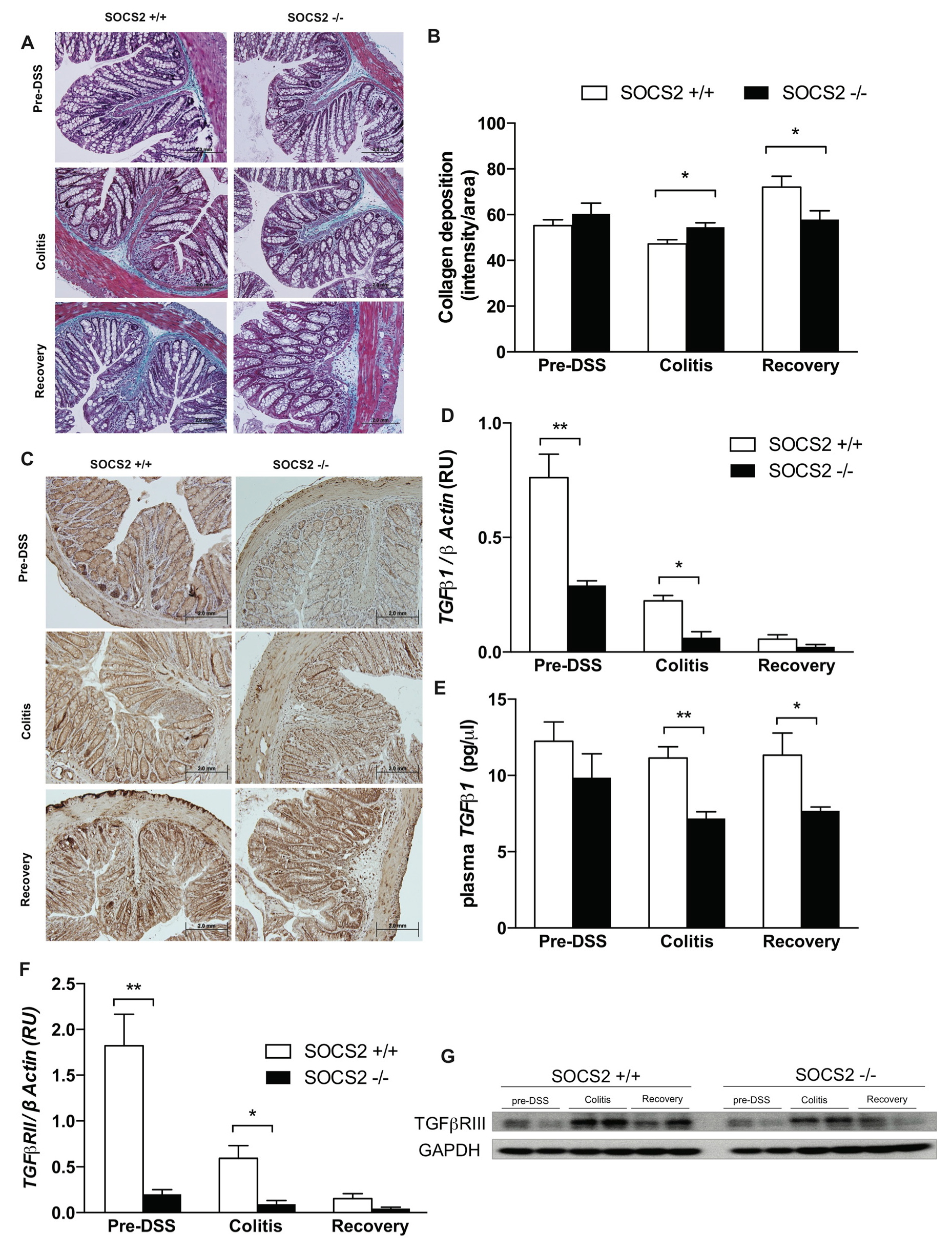
2.3. SOCS2-Deletion Reduces Fibrosis in an Inflammatory Bowel Model
2.4. Fibrosis TGFβ1-Smad Signaling Altered by SOCS2-Deletion
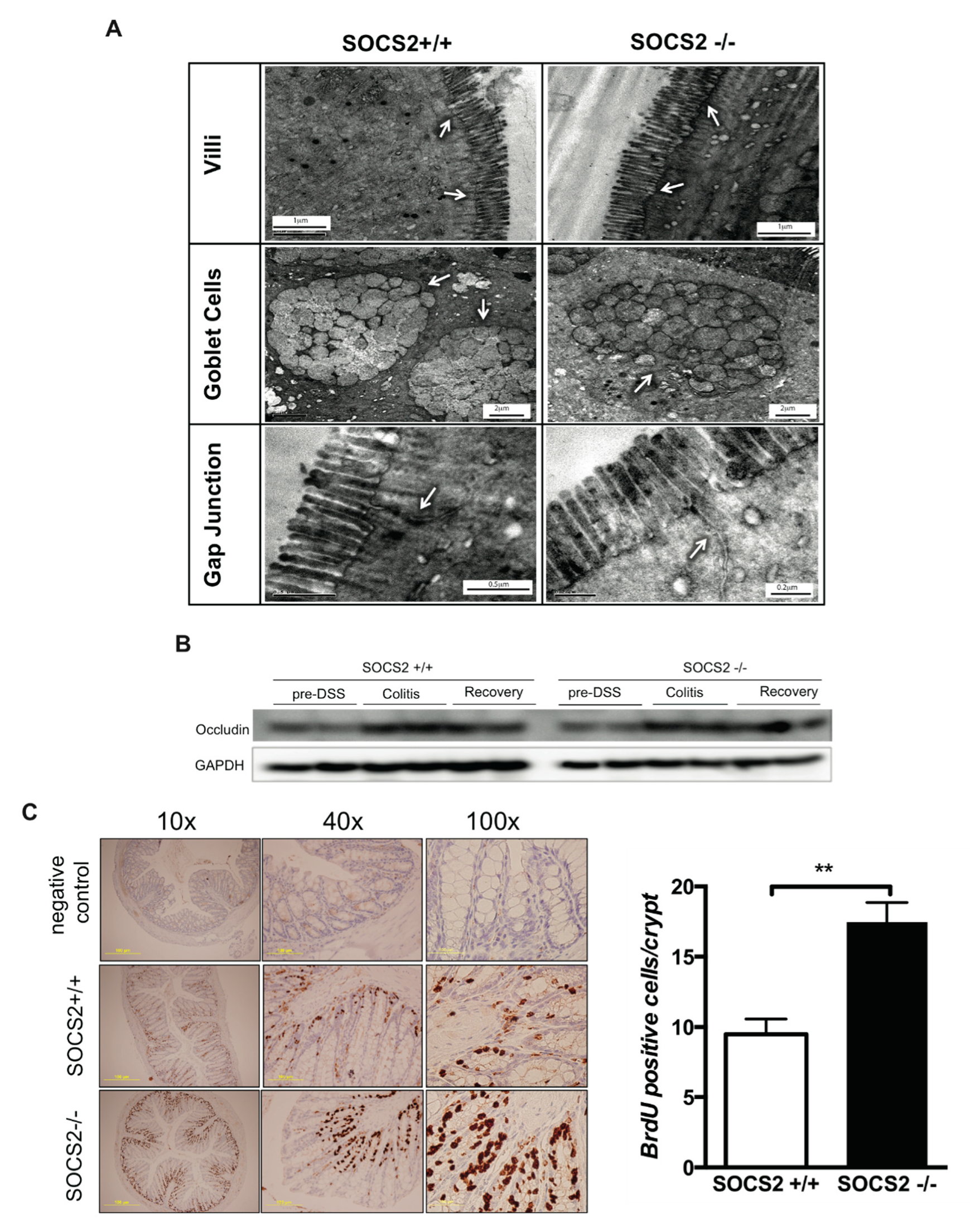
2.5. Role Epithelial Regeneration in Disease Activity
3. Discussion
4. Materials and Methods
4.1. Mice and Induction of Colitis and Recovery
4.2. Histological Evaluation of DSS-Induced Colitis Severity
4.3. Immunohistochemistry
4.4. In Vivo Epithelial Cell Proliferation
4.5. Quantitation of Cytokine Gene Expression Using Real-Time PCR
4.6. Plasma Levels of Cytokines
4.7. Western Blot Analysis
4.8. Transmission Electron Microscopy Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamamoto-Furusho, J.K.; Korzenik, J.R. Crohn’s disease: Innate immunodeficiency? World J. Gastroenterol. 2006, 12, 6751–6755. [Google Scholar] [CrossRef] [PubMed]
- Barahona-Garrido, J.; Hernandez-Calleros, J.; Garcia-Juarez, I.; Yamamoto-Furusho, J.K. Growth factors as treatment for inflammatory bowel disease: A concise review of the evidence toward their potential clinical utility. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2009, 15, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Slonim, A.E.; Bulone, L.; Damore, M.B.; Goldberg, T.; Wingertzahn, M.A.; McKinley, M.J. A preliminary study of growth hormone therapy for Crohn’s disease. New Engl. J. Med. 2000, 342, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Fuller, C.R.; Dieleman, L.A.; DaCosta, C.M.; Haldeman, K.M.; Sartor, R.B.; Lund, P.K. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology 2001, 120, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Sungurtekin, H.; Sungurtekin, U.; Alkanat, M.; Ilkgul, O. The effect of recombinant human growth hormone (rhGH) on trinitrobenzene sulfonic acid-induced colitis in rats: An experimental study. Inflamm. Bowel Dis. 2004, 10, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, E.M.; Li, L.; Hou, Y.T.; Cannon, M.; Christman, G.M.; Bitar, K.N. IGF-I induces collagen and IGFBP-5 mRNA in rat intestinal smooth muscle. Am. J. Physiol. 1997, 273, G875–G882. [Google Scholar] [CrossRef]
- Xin, X.; Hou, Y.T.; Li, L.; Schmiedlin-Ren, P.; Christman, G.M.; Cheng, H.L.; Bitar, K.N.; Zimmermann, E.M. IGF-I increases IGFBP-5 and collagen alpha1(I) mRNAs by the MAPK pathway in rat intestinal smooth muscle cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G777–G783. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.G.; Pucilowska, J.B.; Lund, P.K. Autocrine and paracrine actions of intestinal fibroblast-derived insulin-like growth factors. Am. J. Physiol. 1999, 276, G817–G827. [Google Scholar] [CrossRef]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2012, 10, 639–645. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease: A population-based study. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef]
- Kaneshiro, M.; Melmed, G.Y. Human growth hormone in IBD: Rationale, evidence, and concerns. Pract. Gastroenterol. 2009, 33, 33–36. [Google Scholar]
- Zadjali, F.; Santana-Farre, R.; Vesterlund, M.; Carow, B.; Mirecki-Garrido, M.; Hernandez-Hernandez, I.; Flodstrom-Tullberg, M.; Parini, P.; Rottenberg, M.; Norstedt, G.; et al. SOCS2 deletion protects against hepatic steatosis but worsens insulin resistance in high-fat-diet-fed mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 3282–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadjali, F.; Santana-Farre, R.; Mirecki-Garrido, M.; Ellis, E.; Norstedt, G.; Fernandez-Perez, L.; Flores-Morales, A. Liver X receptor agonist downregulates growth hormone signaling in the liver. Horm. Mol. Biol. Clin. Investig. 2011, 8, 471–478. [Google Scholar] [CrossRef]
- Vesterlund, M.; Zadjali, F.; Persson, T.; Nielsen, M.L.; Kessler, B.M.; Norstedt, G.; Flores-Morales, A. The SOCS2 ubiquitin ligase complex regulates growth hormone receptor levels. PLoS ONE 2011, 6, e25358. [Google Scholar] [CrossRef]
- Alkharusi, A.; Mirecki-Garrido, M.; Ma, Z.; Zadjali, F.; Flores-Morales, A.; Nystrom, T.; Castrillo, A.; Bjorklund, A.; Norstedt, G.; Fernandez-Perez, L. Suppressor of cytokine signaling 2 (SOCS2) deletion protects against multiple low dose streptozotocin-induced type 1 diabetes in adult male mice. Horm. Mol. Biol. Clin. Investig. 2016, 26, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, D.; Greenhalgh, C.J.; Viney, E.; Willson, T.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 2000, 405, 1069–1073. [Google Scholar] [CrossRef]
- Greenhalgh, C.J.; Rico-Bautista, E.; Lorentzon, M.; Thaus, A.L.; Morgan, P.O.; Willson, T.A.; Zervoudakis, P.; Metcalf, D.; Street, I.; Nicola, N.A.; et al. SOCS2 negatively regulates growth hormone action in vitro and in vivo. J. Clin. Investig. 2005, 115, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.E.; Michaylira, C.Z.; Simmons, J.G.; Ney, D.M.; Dahly, E.M.; Heath, J.K.; Lund, P.K. Suppressor of cytokine signaling-2: A growth hormone-inducible inhibitor of intestinal epithelial cell proliferation. Gastroenterology 2004, 127, 570–581. [Google Scholar] [CrossRef]
- Paul, I.; Batth, T.S.; Iglesias-Gato, D.; Al-Araimi, A.; Al-Haddabi, I.; Alkharusi, A.; Norstedt, G.; Olsen, J.V.; Zadjali, F.; Flores-Morales, A. The ubiquitin ligase Cullin5(SOCS2) regulates NDR1/STK38 stability and NF-kappaB transactivation. Sci. Rep. 2017, 7, 42800. [Google Scholar] [CrossRef] [Green Version]
- Theiss, A.L.; Fuller, C.R.; Simmons, J.G.; Liu, B.; Sartor, R.B.; Lund, P.K. Growth hormone reduces the severity of fibrosis associated with chronic intestinal inflammation. Gastroenterology 2005, 129, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Flanders, K.C.; Heine, U.I.; Jakowlew, S.; Kondaiah, P.; Kim, S.J.; Sporn, M.B. Transforming growth factor-beta: Multifunctional regulator of differentiation and development. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1990, 327, 145–154. [Google Scholar]
- Speca, S.; Giusti, I.; Rieder, F.; Latella, G. Cellular and molecular mechanisms of intestinal fibrosis. World J. Gastroenterol. 2012, 18, 3635–3661. [Google Scholar] [CrossRef] [PubMed]
- Caz, V.; Elvira, M.; Tabernero, M.; Grande, A.G.; Lopez-Plaza, B.; de Miguel, E.; Largo, C.; Santamaria, M. Growth Hormone Protects the Intestine Preserving Radiotherapy Efficacy on Tumors: A Short-Term Study. PLoS ONE 2015, 10, e0144537. [Google Scholar] [CrossRef]
- Zadjali, F.; Pike, A.C.; Vesterlund, M.; Sun, J.; Wu, C.; Li, S.S.; Ronnstrand, L.; Knapp, S.; Bullock, A.N.; Flores-Morales, A. Structural basis for c-KIT inhibition by the suppressor of cytokine signaling 6 (SOCS6) ubiquitin ligase. J. Biol. Chem. 2011, 286, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Kazi, J.U.; Sun, J.; Phung, B.; Zadjali, F.; Flores-Morales, A.; Ronnstrand, L. Suppressor of cytokine signaling 6 (SOCS6) negatively regulates Flt3 signal transduction through direct binding to phosphorylated tyrosines 591 and 919 of Flt3. J. Biol. Chem. 2012, 287, 36509–36517. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Debreczeni, J.E.; Edwards, A.M.; Sundstrom, M.; Knapp, S. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2006, 103, 7637–7642. [Google Scholar] [CrossRef] [Green Version]
- Danese, S. Nonimmune cells in inflammatory bowel disease: From victim to villain. Trends Immunol. 2008, 29, 555–564. [Google Scholar] [CrossRef]
- Danese, S. Inflammation and the mucosal microcirculation in inflammatory bowel disease: The ebb and flow. Curr. Opin. Gastroenterol. 2007, 23, 384–389. [Google Scholar] [CrossRef]
- Chidlow, J.H., Jr.; Shukla, D.; Grisham, M.B.; Kevil, C.G. Pathogenic angiogenesis in IBD and experimental colitis: New ideas and therapeutic avenues. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G5–G18. [Google Scholar] [CrossRef] [Green Version]
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in inflammatory bowel disease: Progress in basic and clinical science. Curr. Opin. Gastroenterol. 2008, 24, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in inflammatory bowel disease—Current knowledge and future perspectives. J. Crohn’s Colitis 2008, 2, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucilowska, J.B.; Williams, K.L.; Lund, P.K. Fibrogenesis. I.V. Fibrosis and inflammatory bowel disease: Cellular mediators and animal models. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G653–G659. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Brenmoehl, J.; Leeb, S.; Scholmerich, J.; Rogler, G. Wound healing and fibrosis in intestinal disease. Gut 2007, 56, 130–139. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.G. Regulation of TGF-beta receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.; Cao, Y.; Wang, S.R.; Xu, Y.Z.; Huang, H.; Cui, Y.X.; Huang, Y. Beneficial effect of recombinant human growth hormone on the intestinal mucosa barrier of septic rats. Braz. J. Med. Biol. Res. 2007, 40, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, K.J.; Choi, P.M.; Warner, B.W. The role of growth factors in intestinal regeneration and repair in necrotizing enterocolitis. Semin. Pediatric Surg. 2013, 22, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.; Nivarthi, H.; Mayhew, C.N.; Lo, Y.H.; Noah, T.K.; Vallance, J.; Rulicke, T.; Muller, M.; Jegga, A.G.; Tang, W.; et al. Activated STAT5 confers resistance to intestinal injury by increasing intestinal stem cell proliferation and regeneration. Stem Cell Rep. 2015, 4, 209–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakansson, A.; Branning, C.; Adawi, D.; Molin, G.; Nyman, M.; Jeppsson, B.; Ahrne, S. Blueberry husks, rye bran and multi-strain probiotics affect the severity of colitis induced by dextran sulphate sodium. Scand. J. Gastroenterol. 2009, 44, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. J. Tech. Methods Pathol. 1993, 69, 238–249. [Google Scholar]
- Borody, T.J.; Warren, E.F.; Leis, S.; Surace, R.; Ashman, O. Treatment of ulcerative colitis using fecal bacteriotherapy. J. Clin. Gastroenterol. 2003, 37, 42–47. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Score | Description |
|---|---|---|
| Inflammation severity | 0 | None |
| 1 | Mild | |
| 2 | Moderate | |
| 3 | Severe | |
| Inflammation extent | 0 | None |
| 1 | Mucosa | |
| 2 | Submucosa | |
| 3 | Transmural | |
| Crypt damage | 0 | None |
| 1 | Basal 1⁄3 damaged | |
| 2 | Basal 2⁄3 damaged | |
| 3 | Crypt lost | |
| 4 | Surface epithelial lost | |
| Ulcer | 4 | |
| Lymphocyte infiltration | 3 | |
| Neutrophil infiltration | 2 | |
| Cryptitis | 3 | |
| Crypt abscess | 3 | |
| Edema | 4 | |
| Goblet cell depletion | 3 |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| NOS2 | CATCAACCAGTATTATGGCTC | TTTCCTTTGTTACAGCTTCC |
| IL-1β | GGATGATGATGATAACCTGC | CATGGAGAATATCACTTGTTGG |
| IL-4 | CTGGATTCATCGATAAGCTG | TTTGCATGATGCTCTTTAGG |
| TGFβ1 | CACCGGAGAGCCCTGGATA | TGTACAGCTGCCGCACACA |
| TGFβRI | TGCAATCAGGACCACTGCAATAA | GTGCAATGCAGACGAAGCAGA |
| TGFβRII | AAATTCCCAGCTTCTGGCTCAAC | TGTGCTGTGAGACGGGCTTC |
| β-Actin | GATGTATGAAGGCTTTGGTC | TGTGCACTTTTATTGGTCTC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Araimi, A.; Al Kharusi, A.; Bani Oraba, A.; Al-Maney, M.M.; Al Sinawi, S.; Al-Haddabi, I.; Zadjali, F. Deletion of SOCS2 Reduces Post-Colitis Fibrosis via Alteration of the TGFβ Pathway. Int. J. Mol. Sci. 2020, 21, 3073. https://doi.org/10.3390/ijms21093073
Al-Araimi A, Al Kharusi A, Bani Oraba A, Al-Maney MM, Al Sinawi S, Al-Haddabi I, Zadjali F. Deletion of SOCS2 Reduces Post-Colitis Fibrosis via Alteration of the TGFβ Pathway. International Journal of Molecular Sciences. 2020; 21(9):3073. https://doi.org/10.3390/ijms21093073
Chicago/Turabian StyleAl-Araimi, Amna, Amira Al Kharusi, Asma Bani Oraba, Matar M Al-Maney, Shadia Al Sinawi, Ibrahim Al-Haddabi, and Fahad Zadjali. 2020. "Deletion of SOCS2 Reduces Post-Colitis Fibrosis via Alteration of the TGFβ Pathway" International Journal of Molecular Sciences 21, no. 9: 3073. https://doi.org/10.3390/ijms21093073
APA StyleAl-Araimi, A., Al Kharusi, A., Bani Oraba, A., Al-Maney, M. M., Al Sinawi, S., Al-Haddabi, I., & Zadjali, F. (2020). Deletion of SOCS2 Reduces Post-Colitis Fibrosis via Alteration of the TGFβ Pathway. International Journal of Molecular Sciences, 21(9), 3073. https://doi.org/10.3390/ijms21093073