The Effects of Maternal and Postnatal Dietary Methyl Nutrients on Epigenetic Changes that Lead to Non-Communicable Diseases in Adulthood


{kind=link}
Abstract
:1. Non-Communicable Diseases, Perinatal Diet and Epigenetics
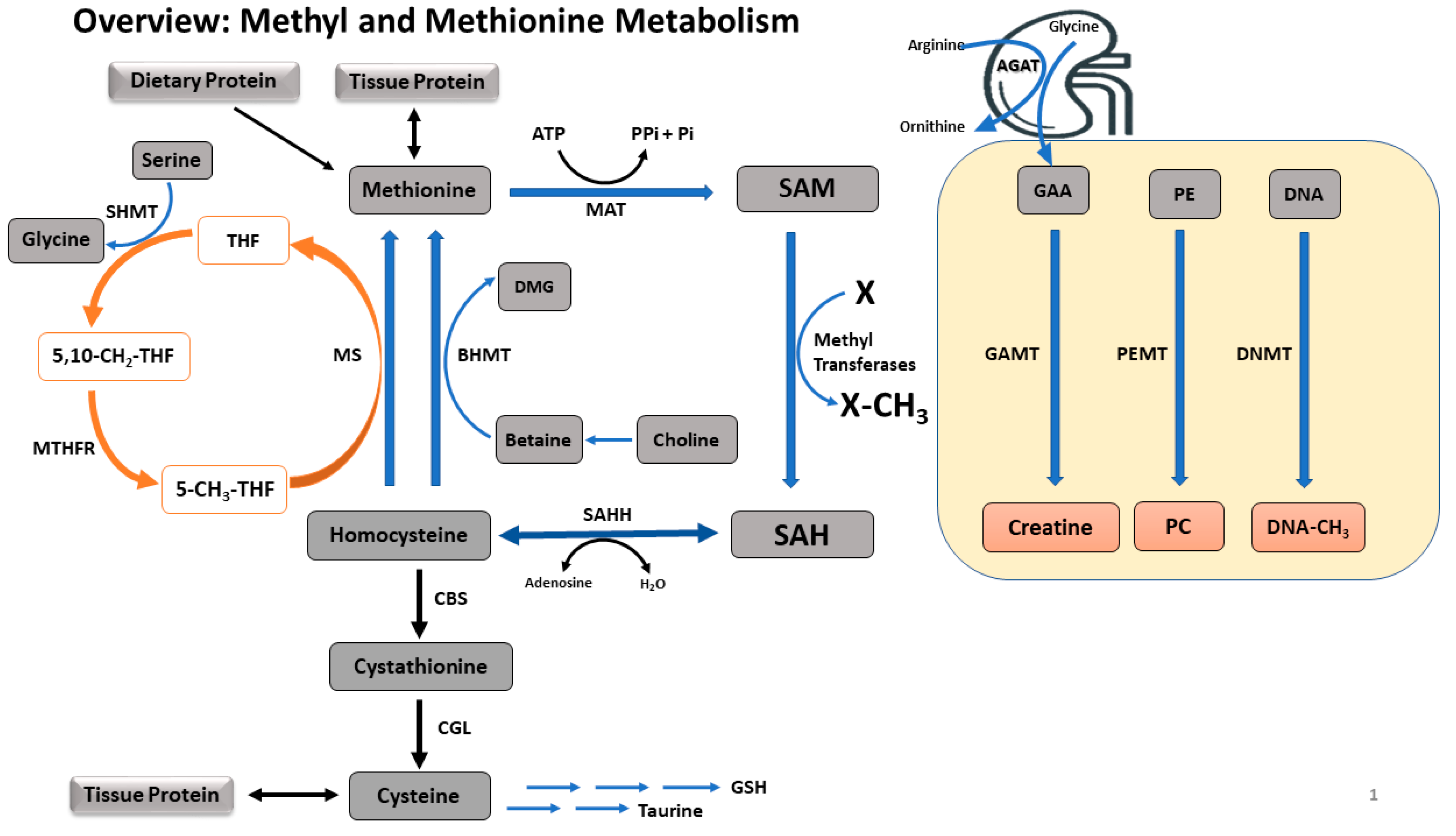
2. Methyl and Methionine Metabolism Pathways
3. Effects of Maternal Dietary Methyl Related Nutrients on Offspring
3.1. Folate
3.2. Choline
3.3. Betaine
3.4. Methionine
4. Effects of Postnatal Dietary Methyl Related Nutrients on Adult Phenotype
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations:
| ACC | Acetyl-CoA carboxylase |
| BHMT | Betaine-homocysteine methyltransferase |
| CBS | Cystathionine beta-synthase |
| 5,10-CH3-THF | 5,10-methylenetetrahydrofolate |
| 5-CH3-THF | 5-methyltetrahydrofolate |
| CGL | Cystathionine gamma-lyase |
| CpG | Cytosine-guanine phosphodiester dinucleotide |
| CYP27α1 | Cholesterol-27a-hydroxylase |
| DOHaD | Developmental origins of health and disease |
| DMG | Dimethylglycine |
| DMR | Differentially methylated region |
| DNA | Deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| FAS | Fatty acid synthase |
| FBP | Fructose-1, 6-bisphosphatase |
| GAA | Guanidinoacetic acid |
| GABAB | Gamma-aminobutyric acid |
| GALK1 | Galactokinase-1 |
| GAMT | Guanidinoacetate methyltransferase |
| GR | Glucocorticoid receptor |
| HMGCR | 3-Hydroxy-3-methyl-glutaryl-coenzyme A reductase |
| IGF | Insulin-like growth factor |
| LDL | Low density lipoprotein |
| LINE-1 | Long interspersed nucleotide element-1 |
| MAT | Methionine adenosyltransferase |
| mRNA | Messenger RNA |
| MS | Methionine synthase |
| MTHFR | Methylenetetrahydrofolate reductase |
| NCD | Non-communicable diseases |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PEMT | Phosphatidylethanolamine methyltransferase |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PyrC | Pyruvate carboxylase |
| RNA | Ribonucleic acid |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| SAHH | S-adenosylhomocystine hydrolase |
| SCD | Stearoyl-CoA desaturase |
| SHMT | Serine hydroxymethyltransferase |
| SREBP1c | Sterol regulatory element-binding protein-1c |
| THF | Tetrahydrofolate |
References
- Darnton-Hill, I.; Nishida, C.; James, W.P. A life course approach to diet, nutrition and the prevention of chronic diseases. Public Health Nutr. 2004, 7, 101–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.R.; Varghese, C. WHO Western Pacific regional action plan for the prevention and control of NCDs (2014–2020). Version 2. Epidemiol. Health 2014, 36, e2014007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, J.E. The nutritional basis of the fetal origins of adult disease. Int. J. Epidemiol. 2001, 30, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, E.; Funtikova, A.N.; Fíto, M.; Schröder, H. Prenatal nutrition and the risk of adult obesity: Long-term effects of nutrition on epigenetic mechanisms regulating gene expression. J. Nutr. Biochem. 2017, 39, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef]
- Vickers, M.H. Early life nutrition, epigenetics and programming of later life disease. Nutrients 2014, 6, 2165–2178. [Google Scholar] [CrossRef]
- Lillycrop, K.A.; Burdge, G.C. Maternal diet as a modifier of offspring epigenetics. J. Dev. Orig. Health Dis. 2015, 6, 88–95. [Google Scholar] [CrossRef] [Green Version]
- McGee, M.; Bainbridge, S.; Fontaine-Bisson, B. A crucial role for maternal dietary methyl donor intake in epigenetic programming and fetal growth outcomes. Nutr. Rev. 2018, 76, 469–478. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chittka, A.; Chittka, L. Epigenetics of royalty. PLoS Biol. 2010, 8, e1000532. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Zhang, X. CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem. Biophys. Res. Commun. 2009, 383, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Trasler, J.M. Epigenetics in spermatogenesis. Mol. Cell Endocrinol. 2009, 306, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Guibert, S.; Forné, T.; Weber, M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012, 22, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.K. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 2011, 6, 838–842. [Google Scholar]
- Tammen, S.A.; Friso, S.; Choi, S.W. Epigenetics: the link between nature and nurture. Mol. Aspects Med. 2013, 34, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Glier, M.B.; Green, T.J.; Devlin, A.M. Methyl nutrients, DNA methylation, and cardiovascular disease. Mol. Nutr. Food Res. 2014, 58, 172–182. [Google Scholar] [CrossRef]
- McBreairty, L.E.; Bertolo, R.F. The dynamics of methionine supply and demand during early development. Appl. Physiol. Nutr. Metab. 2016, 41, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Bertolo, R.F. The pediatric methionine requirement should incorporate remethylation potential and transmethylation demands. Adv. Nutr. 2016, 7, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Chmurzynska, A. Fetal programming: link between early nutrition, DNA methylation, and complex diseases. Nutr. Rev. 2010, 68, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Field, C.J. Early risk determinants and later health outcomes: implications for research prioritization and the food supply. Summary of the workshop. Am. J. Clin. Nutr. 2009, 89, 1533S–1539S. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Dolinoy, D.C.; Lin, J.R.; Smith, C.A.; Shi, X.; Tahiliani, K.G. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 2006, 44, 401–406. [Google Scholar] [CrossRef]
- Cordero, P.; Milagro, F.I.; Campion, J.; Martinez, J.A. Maternal methyl donors supplementation during lactation prevents the hyperhomocysteinemia induced by a high-fat-sucrose intake by dams. Int. J. Mol. Sci. 2013, 14, 24422–24437. [Google Scholar] [CrossRef] [Green Version]
- McBreairty, L.E.; McGowan, R.A.; Brunton, J.A.; Bertolo, R.F. Partitioning of [methyl-3H]methionine to methylated products and protein is altered during high methyl demand conditions in young Yucatan miniature pigs. J. Nutr. 2013, 143, 804–809. [Google Scholar] [CrossRef] [Green Version]
- McBreairty, L.E.; Robinson, J.L.; Furlong, K.R.; Brunton, J.A.; Bertolo, R.F. Guanidinoacetate is more effective than creatine at enhancing tissue creatine stores while consequently limiting methionine availability in Yucatan miniature pigs. PLoS ONE 2015, 10, e0131563. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; McBreairty, L.E.; Randell, E.W.; Brunton, J.A.; Bertolo, R.F. Restriction of dietary methyl donors limits methionine availability and affects the partitioning of dietary methionine for creatine and phosphatidylcholine synthesis in the neonatal piglet. J. Nutr. Biochem. 2016, 35, 81–86. [Google Scholar] [CrossRef]
- Williams, K.T.; Schalinske, K.L. New insights into the regulation of methyl group and homocysteine metabolism. J. Nutr. 2007, 137, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Guéant, J.L.; Elakoum, R.; Ziegler, O.; Coelho, D.; Feigerlova, E.; Daval, J.L.; Guéant-Rodriguez, R.M. Nutritional models of foetal programming and nutrigenomic and epigenomic dysregulations of fatty acid metabolism in the liver and heart. Pflugers Arch. 2014, 466, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Forges, T.; Monnier-Barbarino, P.; Alberto, J.M.; Guéant-Rodriguez, R.M.; Daval, J.L.; Guéant, J.L. Impact of folate and homocysteine metabolism on human reproductive health. Hum. Reprod. Update 2007, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Pellanda, H.; Forges, T.; Bressenot, A.; Chango, A.; Bronowicki, J.P.; Guéant, J.L.; Namour, F. Fumonisin FB1 treatment acts synergistically with methyl donor deficiency during rat pregnancy to produce alterations of H3- and H4-histone methylation patterns in fetuses. Mol. Nutr. Food Res. 2012, 56, 976–995. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.M.; Guéant-Rodriguez, R.M.; Pooya, S.; Brachet, P.; Alberto, J.M.; Jeannesson, E.; Maskali, F.; Gueguen, N.; Marie, P.Y.; Lacolley, P.; et al. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1α by PRMT1 and SIRT1. J. Pathol. 2011, 225, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crott, J.W.; Liu, Z.; Choi, S.W.; Mason, J.B. Folate depletion in human lymphocytes up-regulates p53 expression despite marked induction of strand breaks in exons 5-8 of the gene. Mutat. Res. 2007, 626, 171–179. [Google Scholar] [CrossRef]
- Jacob, R.A.; Gretz, D.M.; Taylor, P.C.; James, S.J.; Pogribny, I.P.; Miller, B.J.; Henning, S.M.; Swendseid, M.E. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J. Nutr. 1998, 128, 1204–1212. [Google Scholar] [CrossRef] [Green Version]
- Fryer, A.A.; Nafee, T.M.; Ismail, K.M.; Carroll, W.D.; Emes, R.D.; Farrell, W.E. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics 2009, 4, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Fraser, A.; Nelson, S.M.; Macdonald-Wallis, C.; Cherry, L.; Butler, E.; Sattar, N.; Lawlor, D.A. Associations of pregnancy complications with calculated cardiovascular disease risk and cardiovascular risk factors in middle age: the Avon Longitudinal Study of Parents and Children. Circulation 2012, 125, 1367–1380. [Google Scholar] [CrossRef]
- Zeisel, S.H. Nutritional importance of choline for brain development. J. Am. Coll. Nutr. 2004, 23, 621S–626S. [Google Scholar] [CrossRef]
- Zeisel, S.H. Importance of methyl donors during reproduction. Am. J. Clin. Nutr. 2009, 89, 673S–677S. [Google Scholar] [CrossRef]
- Zeisel, S. Choline, other methyl-donors and epigenetics. Nutrients 2017, 9, E445. [Google Scholar] [CrossRef]
- Brockington, A.; Lewist, C.; Whartont, S.; Shaw, P.J. Vascular endothelial growth factor and the nervous system. Neuropathol. Appl. Neurobiol. 2004, 30, 427–446. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Craciunescu, C.N.; Zeisel, S.H. Maternal dietary choline deficiency alters angiogenesis in fetal mouse hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 12834–12839. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006, 20, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craciunescu, C.N.; Albright, C.D.; Mar, M.H.; Song, J.; Zeisel, S.H. Choline availability during embryonic development alters progenitor cell mitosis in developing mouse hippocampus. J. Nutr. 2003, 133, 3614–3618. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.M.; Mellott, T.J.; Kovacheva, V.P.; Blusztajn, J.K. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J. Biol. Chem. 2009, 284, 1982–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainfan, E.; Dizik, M.; Stender, M.; Christman, J.K. Rapid appearance of hypomethylated DNA in livers of rats fed cancer-promoting, methyl-deficient diets. Cancer Res. 1989, 49, 4094–4097. [Google Scholar] [PubMed]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.W.; Errington, M.L.; French, P.J.; Fine, A.; Bliss, T.V.; Garel, S.; Charnay, P.; Bozon, B.; Laroche, S.; Davis, S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 2001, 4, 289–296. [Google Scholar] [CrossRef]
- Bozon, B.; Davis, S.; Laroche, S. A requirement for the immediate early gene Zif268 in reconsolidation of recognition memory after retrieval. Neuron 2003, 40, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Mellott, T.J.; Follettie, M.T.; Diesl, V.; Hill, A.A.; Lopez-Coviella, I.; Blusztajn, J.K. Prenatal choline availability modulates hippocampal and cerebral cortical gene expression. FASEB J. 2007, 21, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.C.; Dimova, J.G.; Siddappa, A.J.; Tran, P.V.; Gewirtz, J.C.; Georgieff, M.K. Prenatal choline supplementation ameliorates the long-term neurobehavioral effects of fetal-neonatal iron deficiency in rats. J. Nutr. 2014, 144, 1858–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, S.A. Betaine in human nutrition. Am. J. Clin. Nutr. 2004, 80, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lever, M.; Slow, S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin. Biochem. 2010, 43, 732–744. [Google Scholar] [CrossRef]
- Robinson, J.L.; McBreairty, L.E.; Randell, E.W.; Harding, S.V.; Bartlett, R.K.; Brunton, J.A.; Bertolo, R.F. Betaine or folate can equally furnish remethylation to methionine and increase transmethylation in methionine-restricted neonates. J. Nutr. Biochem. 2018, 59, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Kettunen, H.; Tiihonen, K.; Peuranen, S.; Saarinen, M.T.; Remus, J.C. Dietary betaine accumulates in the liver and intestinal tissue and stabilizes the intestinal epithelial structure in healthy and coccidia-infected broiler chicks. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001, 130, 759–769. [Google Scholar] [CrossRef]
- Fetterer, R.H.; Augustine, P.C.; Allen, P.C.; Barfield, R.C. The effect of dietary betaine on intestinal and plasma levels of betaine in uninfected and coccidia-infected broiler chicks. Parasitol. Res. 2003, 90, 343–348. [Google Scholar] [CrossRef]
- Cai, D.; Jia, Y.; Lu, J.; Yuan, M.; Sui, S.; Song, H.; Zhao, R. Maternal dietary betaine supplementation modifies hepatic expression of cholesterol metabolic genes via epigenetic mechanisms in newborn piglets. Br. J. Nutr. 2014, 112, 1459–1468. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Yuan, M.; Liu, H.; Han, Z.; Pan, S.; Yang, Y.; Zhao, R. Epigenetic and SP1-mediated regulation is involved in the repression of galactokinase 1 gene in the liver of neonatal piglets born to betaine-supplemented sows. Eur. J. Nutr. 2017, 56, 1899–1909. [Google Scholar] [CrossRef]
- Zhao, N.; Yang, S.; Hu, Y.; Dong, H.; Zhao, R. Maternal betaine supplementation in rats induces intergenerational changes in hepatic IGF-1 expression and DNA methylation. Mol. Nutr. Food Res. 2017, 61, 8. [Google Scholar] [CrossRef]
- Knight, L.S.; Piibe, Q.; Lambie, I.; Perkins, C.; Yancey, P. Betaine in the brain: characterization of betaine uptake, its influence on other osmolytes and its potential role in neuroprotection from osmotic stress. Neurochem. Res. 2017, 42, 3490–3503. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, Q.; Li, X.; Cai, D.; Sui, S.; Jia, Y.; Song, H.; Zhao, R. Dietary betaine supplementation to gestational sows enhances hippocampal IGF2 expression in newborn piglets with modified DNA methylation of the differentially methylated regions. Eur. J. Nutr. 2015, 54, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, X.; Jia, Y.; Pan, S.; Li, R.; Yang, X.; Zhao, R. Maternal betaine supplementation during gestation modifies hippocampal expression of GR and its regulatory miRNAs in neonatal piglets. J. Vet. Med. Sci. 2016, 78, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesic, V.; Perovic, M.; Lazic, D.; Kojic, S.; Smiljanic, K.; Ruzdijic, S.; Rakic, L.; Kanazir, S. Long-term intermittent feeding restores impaired GR signaling in the hippocampus of aged rat. J. Steroid Biochem. Mol. Biol. 2015, 149, 43–52. [Google Scholar] [CrossRef]
- McGowan, P.O.; Meaney, M.J.; Szyf, M. Diet and the epigenetic (re)programming of phenotypic differences in behavior. Brain Res. 2008, 1237, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Schwab, U.; Törrönen, A.; Toppinen, L.; Alfthan, G.; Saarinen, M.; Aro, A.; Uusitupa, M. Betaine supplementation decreases plasma homocysteine concentrations but does not affect body weight, body composition, or resting energy expenditure in human subjects. Am. J. Clin. Nutr. 2002, 76, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Wang, J.; Jia, Y.; Liu, H.; Yuan, M.; Dong, H.; Zhao, R. Gestational dietary betaine supplementation suppresses hepatic expression of lipogenic genes in neonatal piglets through epigenetic and glucocorticoid receptor-dependent mechanisms. Biochim. Biophys. Acta 2016, 1861, 41–50. [Google Scholar] [CrossRef]
- Pauwels, S.; Ghosh, M.; Duca, R.C.; Bekaert, B.; Freson, K.; Huybrechts, I.; Langie, S.A.S.; Koppen, G.; Devlieger, R.; Godderis, L. Maternal intake of methyl-group donors affects DNA methylation of metabolic genes in infants. Clin. Epigenetics 2017, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Jacometo, C.B.; Zhou, Z.; Luchini, D.; Corrêa, M.N.; Loor, J.J. Maternal supplementation with rumen-protected methionine increases prepartal plasma methionine concentration and alters hepatic mRNA abundance of 1-carbon, methionine, and transsulfuration pathways in neonatal Holstein calves. J. Dairy Sci. 2017, 100, 3209–3219. [Google Scholar] [CrossRef]
- Jacometo, C.B.; Alharthi, A.S.; Zhou, Z.; Luchini, D.; Loor, J.J. Maternal supply of methionine during late pregnancy is associated with changes in immune function and abundance of microRNA and mRNA in Holstein calf polymorphonuclear leukocytes. J. Dairy Sci. 2018, 101, 8146–8158. [Google Scholar] [CrossRef]
- Liu, G.; Abraham, E. MicroRNAs in immune response and macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacometo, C.B.; Zhou, Z.; Luchini, D.; Trevisi, E.; Corrêa, M.N.; Loor, J.J. Maternal rumen-protected methionine supplementation and its effect on blood and liver biomarkers of energy metabolism, inflammation, and oxidative stress in neonatal Holstein calves. J. Dairy Sci. 2016, 99, 6753–6763. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.D.; Allegrucci, C.; Singh, R.; Gardner, D.S.; Sebastian, S.; Bispham, J.; Thurston, A.; Huntley, J.F.; Rees, W.D.; Maloney, C.A.; et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc. Natl. Acad. Sci. USA 2007, 104, 19351–19356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudicelli, F.; Brabant, A.L.; Grit, I.; Parnet, P.; Amarger, V. Excess of methyl donor in the perinatal period reduces postnatal leptin secretion in rat and interacts with the effect of protein content in diet. PLoS ONE 2013, 8, e68268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, F.; Yan, X.; Yu, Y.; Zhu, X.; Ma, Y.; Yue, Z.; Ou, H.; Yan, Z. Protective effects of maternal methyl donor supplementation on adult offspring of high fat diet-fed dams. J. Nutr. Biochem. 2016, 34, 42–51. [Google Scholar] [CrossRef]
- Cordero, P.; Milagro, F.I.; Campion, J.; Martinez, J.A. Supplementation with methyl donors during lactation to high-fat-sucrose-fed dams protects offspring against liver fat accumulation when consuming an obesogenic diet. J. Dev. Orig. Health Dis. 2014, 5, 385–395. [Google Scholar] [CrossRef]
- Cho, C.E.; Sánchez-Hernández, D.; Reza-López, S.A.; Huot, P.S.; Kim, Y.I.; Anderson, G.H. High folate gestational and post-weaning diets alter hypothalamic feeding pathways by DNA methylation in Wistar rat offspring. Epigenetics 2013, 8, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Langley-Evans, S.C.; Welham, S.J.; Jackson, A.A. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci. 1999, 64, 965–974. [Google Scholar] [CrossRef]
- Plagemann, A.; Harder, T.; Rake, A.; Melchior, K.; Rohde, W.; Dörner, G. Hypothalamic nuclei are malformed in weanling offspring of low protein malnourished rat dams. J. Nutr. 2000, 130, 2582–2589. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Randunu, R.S.; Bertolo, R.F. The Effects of Maternal and Postnatal Dietary Methyl Nutrients on Epigenetic Changes that Lead to Non-Communicable Diseases in Adulthood. Int. J. Mol. Sci. 2020, 21, 3290. https://doi.org/10.3390/ijms21093290
Randunu RS, Bertolo RF. The Effects of Maternal and Postnatal Dietary Methyl Nutrients on Epigenetic Changes that Lead to Non-Communicable Diseases in Adulthood. International Journal of Molecular Sciences. 2020; 21(9):3290. https://doi.org/10.3390/ijms21093290
Chicago/Turabian StyleRandunu, Raniru S., and Robert F. Bertolo. 2020. "The Effects of Maternal and Postnatal Dietary Methyl Nutrients on Epigenetic Changes that Lead to Non-Communicable Diseases in Adulthood" International Journal of Molecular Sciences 21, no. 9: 3290. https://doi.org/10.3390/ijms21093290
APA StyleRandunu, R. S., & Bertolo, R. F. (2020). The Effects of Maternal and Postnatal Dietary Methyl Nutrients on Epigenetic Changes that Lead to Non-Communicable Diseases in Adulthood. International Journal of Molecular Sciences, 21(9), 3290. https://doi.org/10.3390/ijms21093290