New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand?


Abstract
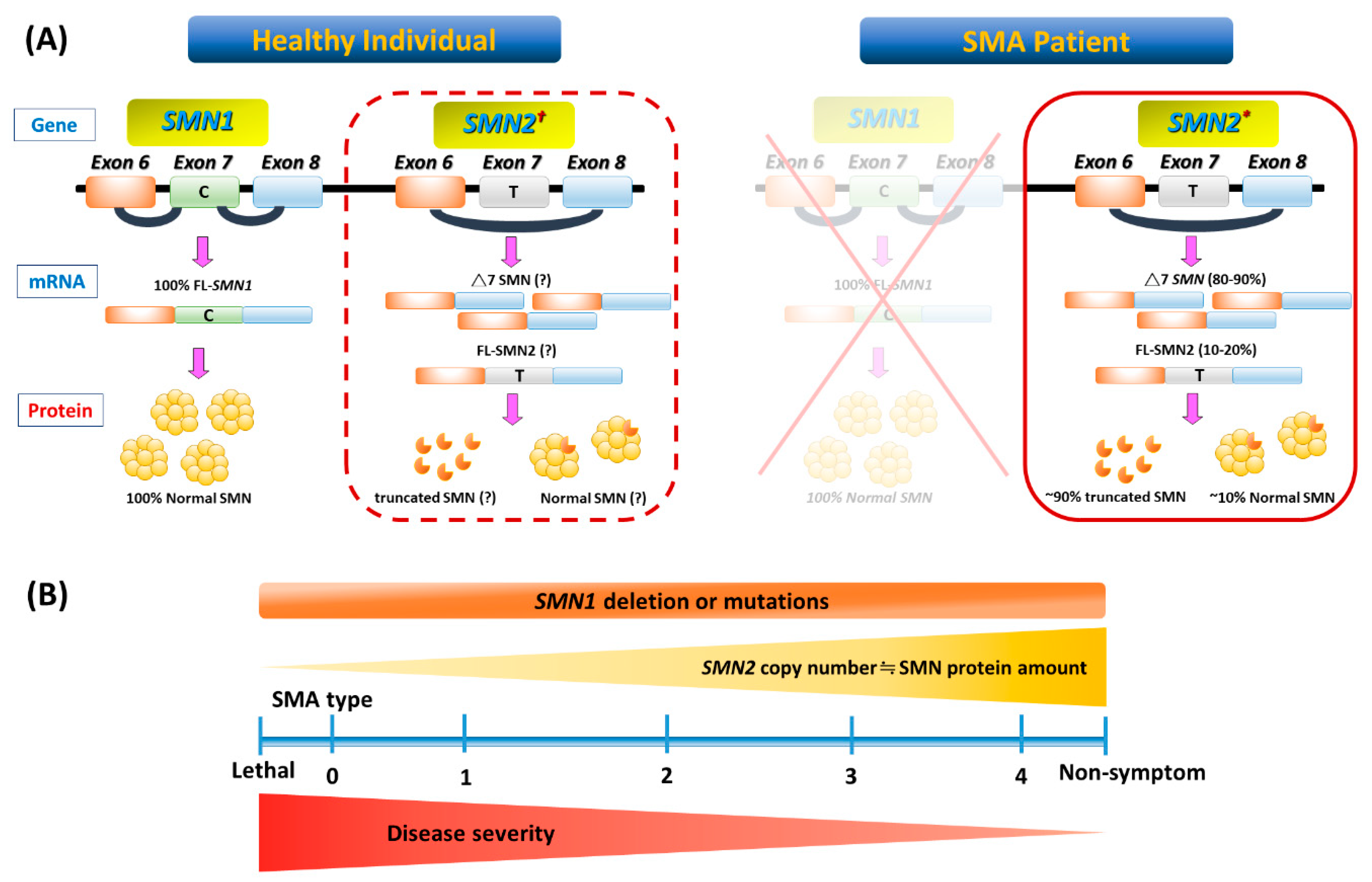
:1. Introduction
2. Clinical Characteristics of SMA
2.1. SMA Phenotypes and Classifications
2.2. The Implication of Phenotypic Classification in SMA Clinical Trials
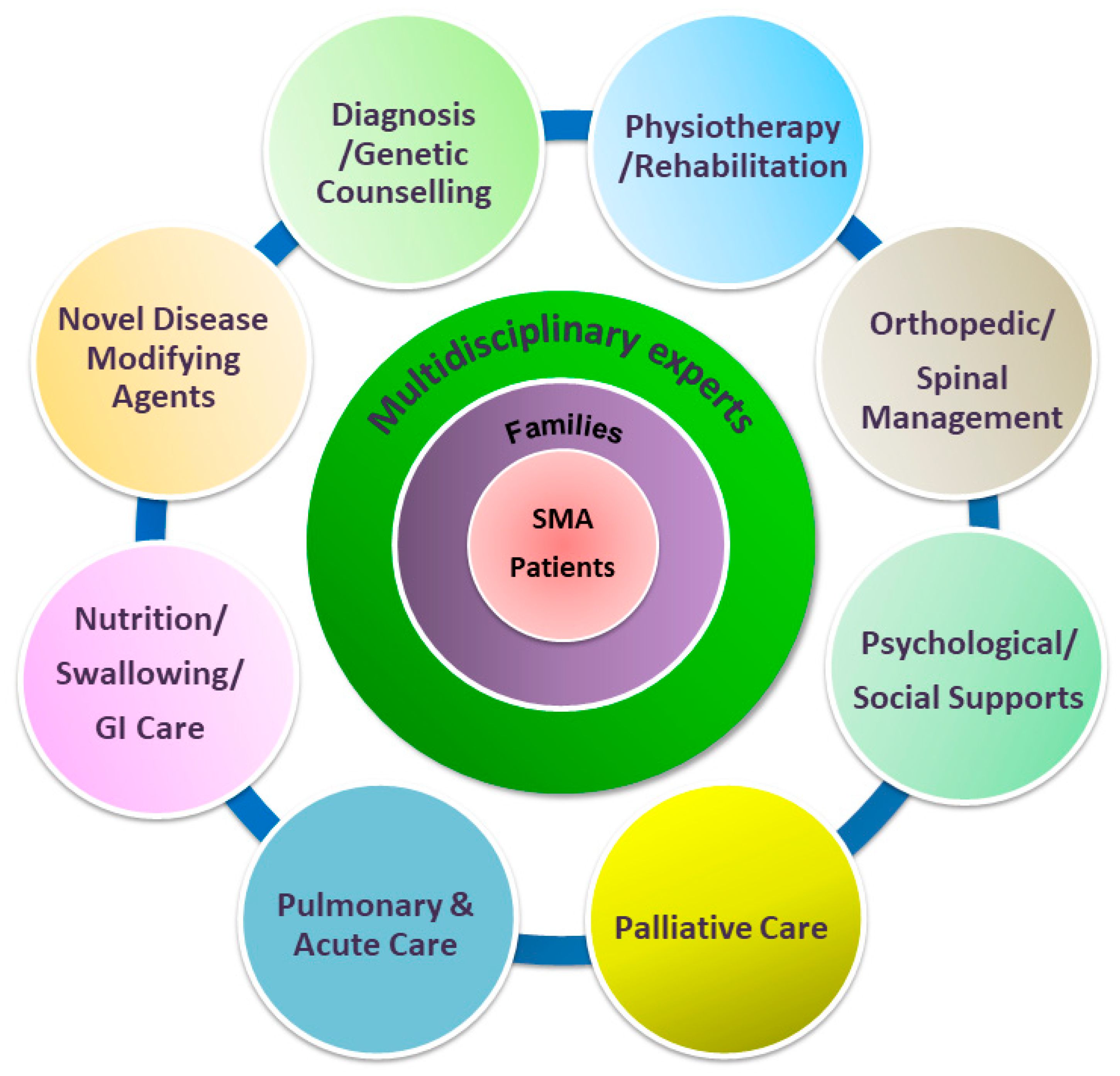
3. Impacts of Evolving Supportive Care in SMA Therapeutic Era
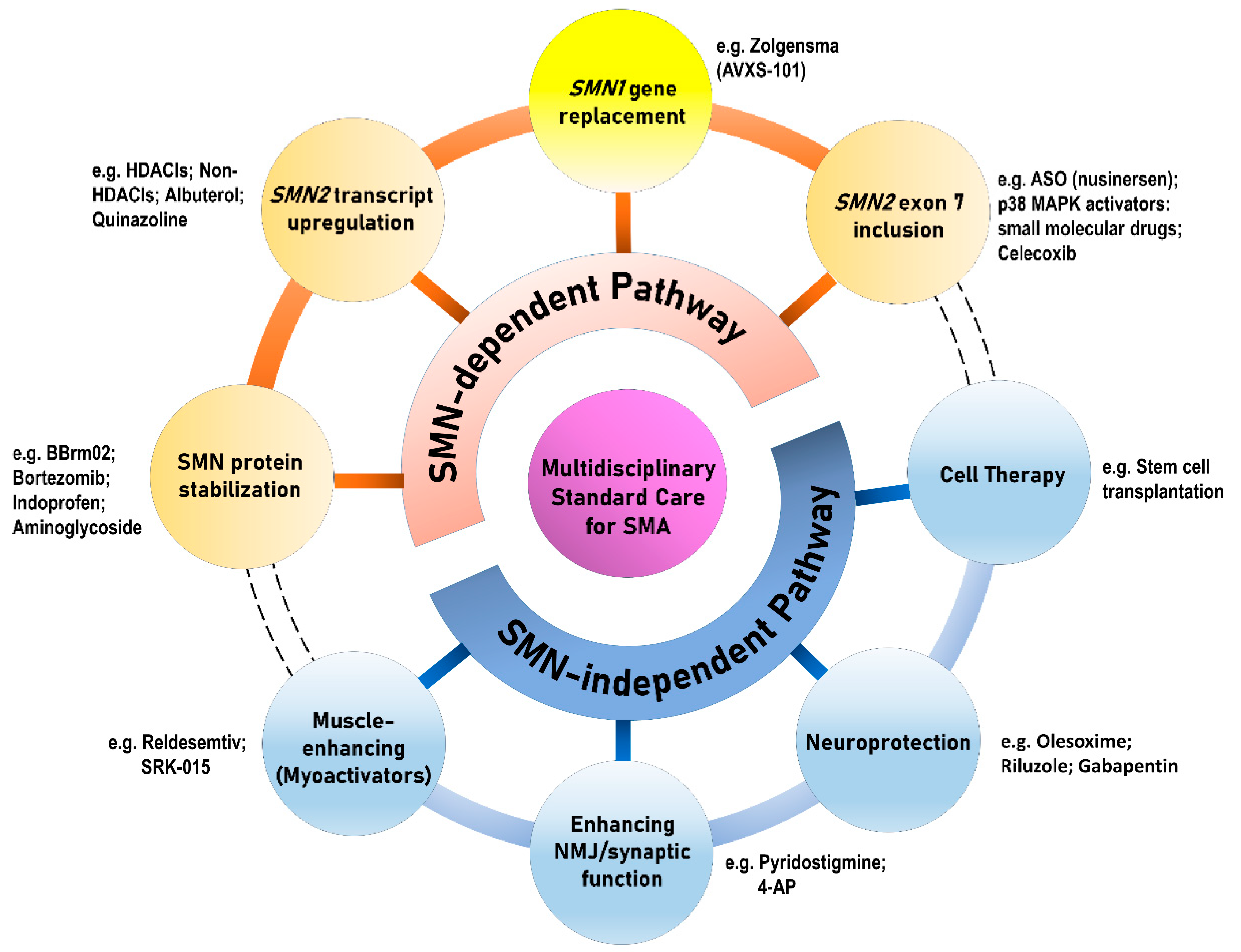
4. Recent Advances in Innovative Therapeutic Approaches for SMA: Focusing SMN and Beyond
5. SMN-Dependent Therapies for SMA
5.1. Previous SMN-Dependent Trials with Indefinable Outcomes
5.2. Nusinersen: The First Approved Splicing-Modify Therapy for SMA
5.3. Gene Therapy for SMA: SMN1 Gene Replacement
5.4. Risdiplam
5.5. Branaplam
5.6. Celecoxib
5.7. Quinazoline
5.8. SMN Protein Stabilizers
6. SMN-Independent Therapies for SMA
6.1. Neuroprotective Agents
6.2. Myostatin Inhibitors
6.3. Skeletal Muscle Troponin Activator: Reldesemtiv
6.4. Agents Targeting Neuromuscular Junction, Synapse, or Neurotransmitter
6.5. Stem Cell Therapy
7. Combination Therapy for SMA
8. Conclusions
Funding
Conflicts of Interest
References
- Darras, B.T. Spinal muscular atrophies. Pediatr. Clin. N. Am. 2015, 62, 743–766. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the smn2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Crawford, T.O.; Paushkin, S.V.; Kobayashi, D.T.; Forrest, S.J.; Joyce, C.L.; Finkel, R.S.; Kaufmann, P.; Swoboda, K.J.; Tiziano, D.; Lomastro, R.; et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e33572. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 4.1–4.31. [Google Scholar]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef]
- Dostie, J.; Mourelatos, Z.; Yang, M.; Sharma, A.; Dreyfuss, G. Numerous microRNPs in neuronal cells containing novel microRNAs. RNA 2003, 9, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabanella, F.; Butchbach, M.E.; Saieva, L.; Carissimi, C.; Burghes, A.H.; Pellizzoni, L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2007, 2, e921. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.M.; Beauvais, A.; Gibeault, S.; Courtney, N.L.; Kothary, R. Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/-) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun. 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.Y.; Simpson, J.E.; Highley, J.R.; Heath, P.R. Spinal muscular atrophy: Factors that modulate motor neurone vulnerability. Neurobiol. Dis. 2017, 102, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindt, H.; Feng, Z.; Mazzasette, C.; Glascock, J.J.; Valdivia, D.; Pyles, N.; Crawford, T.O.; Swoboda, K.J.; Patitucci, T.N.; Ebert, A.D.; et al. Astrocytes influence the severity of spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 4094–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abati, E.; Citterio, G.; Bresolin, N.; Comi, G.P.; Corti, S. Glial cells involvement in spinal muscular atrophy: Could sma be a neuroinflammatory disease? Neurobiol. Dis. 2020, 140, 104870. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bertini, E.; Iannaccone, S.T. Childhood spinal muscular atrophy: Controversies and challenges. Lancet Neurol. 2012, 11, 443–452. [Google Scholar] [CrossRef]
- Nadeau, A.; D’Anjou, G.; Debray, G.; Robitaille, Y.; Simard, L.R.; Vanasse, M. A newborn with spinal muscular atrophy type 0 presenting with a clinicopathological picture suggestive of myotubular myopathy. J. Child. Neurol. 2007, 22, 1301–1304. [Google Scholar] [CrossRef]
- Sansone, V.A.; Racca, F.; Ottonello, G.; Vianello, A.; Berardinelli, A.; Crescimanno, G.; Casiraghi, J.L.; Italian SMA Family Association. 1st Italian SMA Family Association Consensus Meeting: Management and recommendations for respiratory involvement in spinal muscular atrophy (SMA) types I-III, Rome, Italy, 30-31 January 2015. Neuromuscul. Disord. 2015, 25, 979–989. [Google Scholar] [CrossRef] [Green Version]
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; van den Berg, L.H.; van der Pol, W.L. Cardiac pathology in spinal muscular atrophy: A systematic review. Orphanet J. Rare Dis. 2017, 12, 67. [Google Scholar] [CrossRef]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, P.; McDermott, M.P.; Darras, B.T.; Finkel, R.; Kang, P.; Oskoui, M.; Constantinescu, A.; Sproule, D.M.; Foley, A.R.; Yang, M.; et al. Observational study of spinal muscular atrophy type 2 and 3: Functional outcomes over 1 year. Arch. Neurol. 2011, 68, 779–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piepers, S.; van den Berg, L.H.; Brugman, F.; Scheffer, H.; Ruiterkamp-Versteeg, M.; van Engelen, B.G.; Faber, C.G.; de Visser, M.; van der Pol, W.L.; Wokke, J.H. A natural history study of late onset spinal muscular atrophy types 3b and 4. J. Neurol. 2008, 255, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; McDermott, M.P.; Kaufmann, P.; Darras, B.T.; Chung, W.K.; Sproule, D.M.; Kang, P.B.; Foley, A.R.; Yang, M.L.; Martens, W.B.; et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014, 83, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Lucibello, S.; Perulli, M.; Coratti, G.; de Sanctis, R.; Pera, M.C.; Pane, M.; Montes, J.; de Vivo, D.C.; Darras, B.T.; et al. Longitudinal natural history of type I spinal muscular atrophy: A critical review. Orphanet J. Rare Dis. 2020, 15, 84. [Google Scholar] [CrossRef]
- Rudnik-Schoneborn, S.; Berg, C.; Zerres, K.; Betzler, C.; Grimm, T.; Eggermann, T.; Eggermann, K.; Wirth, R.; Wirth, B.; Heller, R. Genotype-phenotype studies in infantile spinal muscular atrophy (SMA) type I in Germany: Implications for clinical trials and genetic counselling. Clin. Genet. 2009, 76, 168–178. [Google Scholar] [CrossRef]
- Tizzano, E.F.; Finkel, R.S. Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscul. Disord. 2017, 27, 883–889. [Google Scholar] [CrossRef]
- Sansone, V.A.; Pirola, A.; Albamonte, E.; Pane, M.; Lizio, A.; D’Amico, A.; Catteruccia, M.; Cutrera, R.; Bruno, C.; Pedemonte, M.; et al. Respiratory Needs in Patients with Type 1 Spinal Muscular Atrophy Treated with Nusinersen. J. Pediatr. 2020, 219, 223–228.e4. [Google Scholar] [CrossRef]
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.A.; Turner, B.J.; Gillingwater, T.H.; Swoboda, K.J.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2017, 81, 355–368. [Google Scholar] [CrossRef]
- Wang, C.H.; Finkel, R.S.; Bertini, E.S.; Schroth, M.; Simonds, A.; Wong, B.; Aloysius, A.; Morrison, L.; Main, M.; Crawford, T.O.; et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child. Neurol. 2007, 22, 1027–1049. [Google Scholar] [CrossRef]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, B.G.; Crawford, T.O.; Sumner, C.J. Emerging treatment options for spinal muscular atrophy. Curr. Treat. Options Neurol. 2009, 11, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.G.; Hsieh-Li, H.M.; Jong, Y.J.; Wang, N.M.; Tsai, C.H.; Li, H. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. USA 2001, 98, 9808–9813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreml, J.; Riessland, M.; Paterno, M.; Garbes, L.; Rossbach, K.; Ackermann, B.; Kramer, J.; Somers, E.; Parson, S.H.; Heller, R.; et al. Severe sma mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet. 2013, 21, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Somers, E.; Lees, R.D.; Hoban, K.; Sleigh, J.N.; Zhou, H.; Muntoni, F.; Talbot, K.; Gillingwater, T.H.; Parson, S.H. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann. Neurol. 2016, 79, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Deguise, M.O.; De Repentigny, Y.; McFall, E.; Auclair, N.; Sad, S.; Kothary, R. Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum. Mol. Genet. 2017, 26, 801–819. [Google Scholar] [CrossRef] [Green Version]
- Nery, F.C.; Siranosian, J.J.; Rosales, I.; Deguise, M.O.; Sharma, A.; Muhtaseb, A.W.; Nwe, P.; Johnstone, A.J.; Zhang, R.; Fatouraei, M.; et al. Impaired kidney structure and function in spinal muscular atrophy. Neurol. Genet. 2019, 5, e353. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 2020, 1, CD006282. [Google Scholar] [CrossRef]
- Perego, M.G.L.; Galli, N.; Nizzardo, M.; Govoni, A.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Cell. Mol. Life Sci. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Lunke, S.; El-Osta, A. Applicability of histone deacetylase inhibition for the treatment of spinal muscular atrophy. Neurotherapeutics 2013, 10, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst. Rev. 2019, 12, CD006281. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, J.; Zabidi-Hussin, Z.A.; Sasongko, T.H. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet. Mol. Biol. 2013, 36, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Calder, A.N.; Androphy, E.J.; Hodgetts, K.J. Small Molecules in Development for the Treatment of Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 10067–10083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Grzeschik, S.M.; Ganta, M.; Prior, T.W.; Heavlin, W.D.; Wang, C.H. Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann. Neurol. 2005, 58, 194–202. [Google Scholar] [CrossRef]
- Liang, W.C.; Yuo, C.Y.; Chang, J.G.; Chen, Y.C.; Chang, Y.F.; Wang, H.Y.; Ju, Y.H.; Chiou, S.S.; Jong, Y.J. The effect of hydroxyurea in spinal muscular atrophy cells and patients. J. Neurol. Sci. 2008, 268, 87–94. [Google Scholar] [CrossRef]
- Chen, T.H.; Chang, J.G.; Yang, Y.H.; Mai, H.H.; Liang, W.C.; Wu, Y.C.; Wang, H.Y.; Huang, Y.B.; Wu, S.M.; Chen, Y.C.; et al. Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy. Neurology 2010, 75, 2190–2197. [Google Scholar] [CrossRef]
- Angelozzi, C.; Borgo, F.; Tiziano, F.D.; Martella, A.; Neri, G.; Brahe, C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J. Med. Genet. 2008, 45, 29–31. [Google Scholar] [CrossRef]
- Kinali, M.; Mercuri, E.; Main, M.; De Biasia, F.; Karatza, A.; Higgins, R.; Banks, L.M.; Manzur, A.Y.; Muntoni, F. Pilot trial of albuterol in spinal muscular atrophy. Neurology 2002, 59, 609–610. [Google Scholar] [CrossRef] [PubMed]
- Pane, M.; Staccioli, S.; Messina, S.; D’Amico, A.; Pelliccioni, M.; Mazzone, E.S.; Cuttini, M.; Alfieri, P.; Battini, R.; Main, M.; et al. Daily salbutamol in young patients with SMA type II. Neuromuscul. Disord. 2008, 18, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Yang, Y.H.; Mai, H.H.; Liang, W.C.; Wu, Y.C.; Wang, H.Y.; Jong, Y.J. Reliability and validity of outcome measures of in-hospital and at-home visits in a randomized, double-blind, placebo-controlled trial for spinal muscular atrophy. J. Child. Neurol. 2014, 29, 1680–1684. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Shishimorova, M.; Cao, L.C.; Gangwani, L.; Singh, R.N. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009, 6, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of enhanced survival motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Invest. 2014, 124, 785–800. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Dangouloff, T.; Burghes, A.; Tizzano, E.F.; Servais, L.; NBS SMA Study Group. 244th ENMC international workshop: Newborn screening in spinal muscular atrophy May 10-12, 2019, Hoofdorp, The Netherlands. Neuromuscul. Disord. 2020, 30, 93–103. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.T.; Russell, J.S.; Wiley, V.; Alexander, I.E.; Farrar, M.A. The implementation of newborn screening for spinal muscular atrophy: The Australian experience. Genet. Med. 2020, 22, 557–565. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Gidaro, T.; Servais, L. Nusinersen treatment of spinal muscular atrophy: Current knowledge and existing gaps. Dev. Med. Child. Neurol. 2019, 61, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Tizzano, E.F. The clinical landscape for SMA in a new therapeutic era. Gene. Ther. 2017, 24, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Nash, L.A.; Burns, J.K.; Chardon, J.W.; Kothary, R.; Parks, R.J. Spinal Muscular Atrophy: More than a Disease of Motor Neurons? Curr. Mol. Med. 2016, 16, 779–792. [Google Scholar] [CrossRef]
- Kim, J.K.; Jha, N.N.; Feng, Z.; Faleiro, M.R.; Chiriboga, C.A.; Wei-Lapierre, L.; Dirksen, R.T.; Ko, C.P.; Monani, U.R. Muscle-specific smn reduction reveals motor neuron-independent disease in spinal muscular atrophy models. J. Clin. Investig. 2020, 130, 1271–1287. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Hua, Y.; Liu, Y.H.; Sahashi, K.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015, 29, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Al-Zaidy, S.A.; Mendell, J.R. From Clinical Trials to Clinical Practice: Practical Considerations for Gene Replacement Therapy in SMA Type 1. Pediatr. Neurol. 2019, 100, 3–11. [Google Scholar] [CrossRef]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants With SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Shell, R.; Day, J.W.; Chiriboga, C.A.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Lannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; et al. Onasemnogene Abeparvovec Gene-Replacement Therapy for Spinal Muscular Atrophy Type 1: Pivotal Phase 3 Study (STR1VE) Update. In Proceedings of the CureSMA 23rd Annual SMA Researcher Meeting, Anaheim, CA, USA, 28–30 June 2019. [Google Scholar]
- Novartis. Novartis Announces AVXS-101 Intrathecal Study Update. Available online: https://www.novartis.com/news/media-releases/novartis-announces-avxs-101-intrathecal-study-update (accessed on 30 March 2020).
- van der Ploeg, A.T. The Dilemma of Two Innovative Therapies for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1786–1787. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Yue, Y.; Zhang, K.; Hakim, C.H.; Kodippili, K.; McDonald, T.; Duan, D. AAV-8 is more efficient than AAV-9 in transducing neonatal dog heart. Hum. Gene Ther. Methods 2015, 26, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Molecul. Ther. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef]
- Kletzl, H.; Marquet, A.; Gunther, A.; Tang, W.; Heuberger, J.; Groeneveld, G.J.; Birkhoff, W.; Mercuri, E.; Lochmuller, H.; Wood, C.; et al. The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: Results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Sturm, S.; Gunther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Seabrook, T.; Baranello, G.; Servais, L.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson1, R.; Kletzl11, H.; et al. FIREFISH part 1: Early clinical results following an increase of SMN protein in infants with type 1 spinal muscular atrophy (SMA) treated with Risdiplam (RG7916). In Proceedings of the Communication Presented at MDA Clinical & Scientific Conference, Orlando, FL, USA, 13–17 April 2019. [Google Scholar]
- Roche. Roche’s Risdiplam Meets Primary Endpoint in Pivotal SUNFISH Trial in People with Type 2 or 3 Spinal Muscular Atrophy. Available online: https://www.roche.com/media/releases/med-cor-2019-11-11.htm (accessed on 16 March 2019).
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef] [PubMed]
- Jevtic, S.; Carr, D.; Dobrzycka-Ambrozevicz, A. Branaplam in Type 1 spinal muscular atrophy: Second part of a phase I/II Study. In Proceedings of the CureSMA 23rd Annual SMA Researcher Meeting, Anaheim, CA, USA, 28–30 June 2019. [Google Scholar]
- Farooq, F.; Abadia-Molina, F.; MacKenzie, D.; Hadwen, J.; Shamim, F.; O’Reilly, S.; Holcik, M.; MacKenzie, A. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum. Mol. Genet. 2013, 22, 3415–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarecki, J.; Chen, X.; Bernardino, A.; Coovert, D.D.; Whitney, M.; Burghes, A.; Stack, J.; Pollok, B.A. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: Early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 2005, 14, 2003–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogliotti, R.G.; Cardona, H.; Singh, J.; Bail, S.; Emery, C.; Kuntz, N.; Jorgensen, M.; Durens, M.; Xia, B.; Barlow, C.; et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 2013, 22, 4084–4101. [Google Scholar] [CrossRef] [Green Version]
- Jedrzejowska, M.; Kostera-Pruszczyk, A. Spinal muscular atrophy - new therapies, new challenges. Neurol. Neurochir. Polska 2020, 54, 8–13. [Google Scholar] [CrossRef]
- Van Meerbeke, J.; Gibbs, R.; Plasterer, H.; Feng, Z.; Lin, M.-Y.; Wee, C.; Xia, B.; Jacques, V.; Rusche, J.; Ko, C.-P.; et al. The Therapeutic Effects of RG3039 in Severe Spinal Muscular Atrophy Mice and Normal Human Volunteers (S25.003). Neurology 2012, 78, S25. [Google Scholar] [CrossRef]
- Heier, C.R.; DiDonato, C.J. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum. Mol. Genet. 2009, 18, 1310–1322. [Google Scholar] [CrossRef] [Green Version]
- Cobb, M.S.; Rose, F.F.; Rindt, H.; Glascock, J.J.; Shababi, M.; Miller, M.R.; Osman, E.Y.; Yen, P.F.; Garcia, M.L.; Martin, B.R.; et al. Development and characterization of an SMN2-based intermediate mouse model of Spinal Muscular Atrophy. Hum. Mol. Genet. 2013, 22, 1843–1855. [Google Scholar] [CrossRef]
- Greif, H.; Rosin-Arbesfeld, R.; Megiddo, D. BBrm2, A Read-through Repurposed Drug, Shows Proof of Efficacy in SMA Treatment. In Proceedings of the Cure SMA 19th Annual SMA Researcher Meeting, Kansas City, MO, USA, 18−20 June 2015. [Google Scholar]
- Kwon, D.Y.; Motley, W.W.; Fischbeck, K.H.; Burnett, B.G. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum. Mol. Genet. 2011, 20, 3667–3677. [Google Scholar] [CrossRef] [Green Version]
- Kaifer, K.A.; Villalon, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.D.W.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef] [Green Version]
- Sumner, C.J.; Crawford, T.O. Two breakthrough gene-targeted treatments for spinal muscular atrophy: Challenges remain. J. Clin. Investig. 2018, 128, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, E.; Dessaud, E.; Mercuri, E.; Muntoni, F.; Kirschner, J.; Reid, C.; Lusakowska, A.; Comi, G.P.; Cuisset, J.M.; Abitbol, J.L.; et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 513–522. [Google Scholar] [CrossRef]
- Haddad, H.; Cifuentes-Diaz, C.; Miroglio, A.; Roblot, N.; Joshi, V.; Melki, J. Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve 2003, 28, 432–437. [Google Scholar] [CrossRef]
- Merlini, L.; Solari, A.; Vita, G.; Bertini, E.; Minetti, C.; Mongini, T.; Mazzoni, E.; Angelini, C.; Morandi, L. Role of gabapentin in spinal muscular atrophy: Results of a multicenter, randomized Italian study. J. Child. Neurol. 2003, 18, 537–541. [Google Scholar] [CrossRef]
- Pirruccello-Straub, M.; Jackson, J.; Wawersik, S.; Webster, M.T.; Salta, L.; Long, K.; McConaughy, W.; Capili, A.; Boston, C.; Carven, G.J.; et al. Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci. Rep. 2018, 8, 2292. [Google Scholar] [CrossRef]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef]
- Liu, M.; Hammers, D.W.; Barton, E.R.; Sweeney, H.L. Activin Receptor Type IIB Inhibition Improves Muscle Phenotype and Function in a Mouse Model of Spinal Muscular Atrophy. PLoS ONE 2016, 11, e0166803. [Google Scholar] [CrossRef]
- AliveGen. R&D Pipeline: ALG-801. Available online: http://www.alivegen.com/r-d-pipeline (accessed on 22 April 2020).
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Chyung, Y. Interim Results from a Phase 1 Study of SRK-015, a Fully Human Monoclonal Antibody that Inhibits Myostatin Activation. In Proceedings of the CureSMA 23rd Annual SMA Researcher Meeting, Anaheim, CA, USA, 28–30 June 2019. [Google Scholar]
- Hwee, D.T.; Kennedy, A.R.; Hartman, J.J.; Ryans, J.; Durham, N.; Malik, F.I.; Jasper, J.R. The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J. Pharmacol. Exp. Ther. 2015, 353, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Andrews, J.A.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F.I. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W. Update of CY 5021: A Phase 2 Clinical Trial of Reldesemtiv, a Fast Skeletal Muscle Troponin Activator (FSTA), for the Potential Treatment of Spinal Muscular Atrophy. In Proceedings of the CureSMA 22nd Annual SMA Researcher Meeting, Dallas, TX, USA, 14–16 June 2018. [Google Scholar]
- Wadman, R.I.; Vrancken, A.F.; van den Berg, L.H.; van der Pol, W.L. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Stam, M.; Wadman, R.I.; Wijngaarde, C.A.; Bartels, B.; Asselman, F.L.; Otto, L.A.M.; Goedee, H.S.; Habets, L.E.; de Groot, J.F.; Schoenmakers, M.; et al. Protocol for a phase II, monocentre, double-blind, placebo-controlled, cross-over trial to assess efficacy of pyridostigmine in patients with spinal muscular atrophy types 2-4 (SPACE trial). BMJ Open 2018, 8, e019932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imlach, W.L.; Beck, E.S.; Choi, B.J.; Lotti, F.; Pellizzoni, L.; McCabe, B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell 2012, 151, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, F.; De Vita, D.; Bortolami, M.; Coluccia, A.; Di Santo, R.; Costi, R.; Andrisano, V.; Alabiso, F.; Bergamini, C.; Fato, R.; et al. New pyridine derivatives as inhibitors of acetylcholinesterase and amyloid aggregation. Eur. J. Med. Chem. 2017, 141, 197–210. [Google Scholar] [CrossRef]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Corti, S.; Nizzardo, M.; Nardini, M.; Donadoni, C.; Salani, S.; Ronchi, D.; Saladino, F.; Bordoni, A.; Fortunato, F.; Del Bo, R.; et al. Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J. Clin. Investig. 2008, 118, 3316–3330. [Google Scholar] [CrossRef]
- Corti, S.; Nizzardo, M.; Nardini, M.; Donadoni, C.; Salani, S.; Ronchi, D.; Simone, C.; Falcone, M.; Papadimitriou, D.; Locatelli, F.; et al. Embryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in mice. Brain 2010, 133, 465–481. [Google Scholar] [CrossRef] [Green Version]
- Poletti, A.; Fischbeck, K.H. Combinatorial treatment for spinal muscular atrophy: An Editorial for ’Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in Spinal Muscular Atrophy cells’ on page 264. J. Neurochem. 2020, 153, 146–149. [Google Scholar]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachexia Sarcopenia Muscle 2020, in press. [Google Scholar] [CrossRef]
- Lee, B.H.; Collins, E.; Lewis, L.; Guntrum, D.; Eichinger, K.; Voter, K.; Abdel-Hamid, H.Z.; Ciafaloni, E. Combination therapy with nusinersen and AVXS-101 in SMA type 1. Neurology 2019, 93, 640–641. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| SMA Type (Historical Name) | OMIM | Onset Age | Motor Milestones Achieved | Subclassification | Natural History | Other Features | Estimated SMN2 Copies | Estimated SMA Proportion |
|---|---|---|---|---|---|---|---|---|
| Type 0 | - | Prenatal or at birth | Never sits, never head control | - | Death < 1 mo if untreated | Joint contractures, cardiac defect, facial diplegia, immediate respiratory failure after birth | 1 SMN2 copy in ~100% of patients | Unclear, Maybe < 1% |
| Type 1 (Werdnig-Hoffmann disease) | 253300 | 0–6 mo | Never sits, some achieve head control | 1A: Onset < 1 mo, usually by 2 wk; head control absent 1B: Onset 1–3 mo; poor or absent head control 1C: Onset 3–6 mo; head control achieved | 1A: Death < 6 mo if untreated 1B and 1C: death < 2 yr if untreated | 1A: Very similar to type 0 SMA 1B and 1C: Tongue fasciculation, swallowing difficulties, early respiratory failure | 1 or 2 SMN2 Copies in ~80% of patients | ~60% |
| Type 2 (Dubowitz disease) | 253550 | 7–18 mo | Sits but never stands | 2A: Sits independently, may lose the ability to sit in later life 2B: Sits independently, maintains the ability to sit According to functional level, decimal classification ranging from 2.1 to 2.9 | Usually survive >2 yr; ~70% alive at 25 yr | Proximal weakness, postural hand tremor, normal intellectual ability, kyphoscoliosis | 3 SMN2 copies in >70% patients | ~27% |
| Type 3 (Kugelberg–Welander disease) | 253400 | >18 mo | Stands and walks | 3A: Onset between 18 and 36 mo 3B: Onset >3 yr | Survival into adulthood | May have hand tremor, resembles muscular dystrophy 3A: Scoliosis, usually early loss of ambulation | 3 or 4 SMN2 copies in ~95% of patients | ~12% |
| Type 4 | 271150 | 10–30 yr, usually >21 yr | Stands and walks | - | Survival into adulthood | Usually preserved walking ability | 4 or more SMN2 copies in >90% | ~1% |
| Therapeutic Pathways | Pathologic Points | Therapeutic Targets | Therapeutic Agents | Trial Status (Completed or Ongoing)/Results |
|---|---|---|---|---|
| SMN-dependent | SMN1 mutation | SMN1 replacement | Zolgensma (AVXS-101) | FDA-Approved |
| Alternative splicing of SMN2 mRNA | Promote exon 7 inclusion |
|
| |
| Decreased full length SMN mRNA | Upregulation of SMN2 transcript |
|
| |
| SMN protein degradation | Stabilizing SMN protein |
| All are preclinical | |
| SMN-independent | Anabolic abnormalities | Muscle-enhancing agent (Myoactivators) |
|
|
| Neuromuscular junction defect | Enhancing neurotransmitters |
|
| |
| Motor neuron loss | Neuroprotection |
|
| |
| Cell therapy for neurotrophic support | Stem cells | Preclinical |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. https://doi.org/10.3390/ijms21093297
Chen T-H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? International Journal of Molecular Sciences. 2020; 21(9):3297. https://doi.org/10.3390/ijms21093297
Chicago/Turabian StyleChen, Tai-Heng. 2020. "New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand?" International Journal of Molecular Sciences 21, no. 9: 3297. https://doi.org/10.3390/ijms21093297
APA StyleChen, T. -H. (2020). New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? International Journal of Molecular Sciences, 21(9), 3297. https://doi.org/10.3390/ijms21093297