Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery

Abstract
:1. Introduction
2. A CDK-Centered Overview of RNAPII Transcriptional Regulation
3. Catalytic Mechanism of RNAPII Transcription
4. CDK Inhibitors Affecting RNAPII Transcription
4.1. Pan-CDK Inhibitors
4.1.1. Flavopiridol
4.1.2. Roscovitine (Seliciclib)
4.1.3. SNS-032
4.1.4. Dinaciclib
4.2. Selective CDK Inhibitors
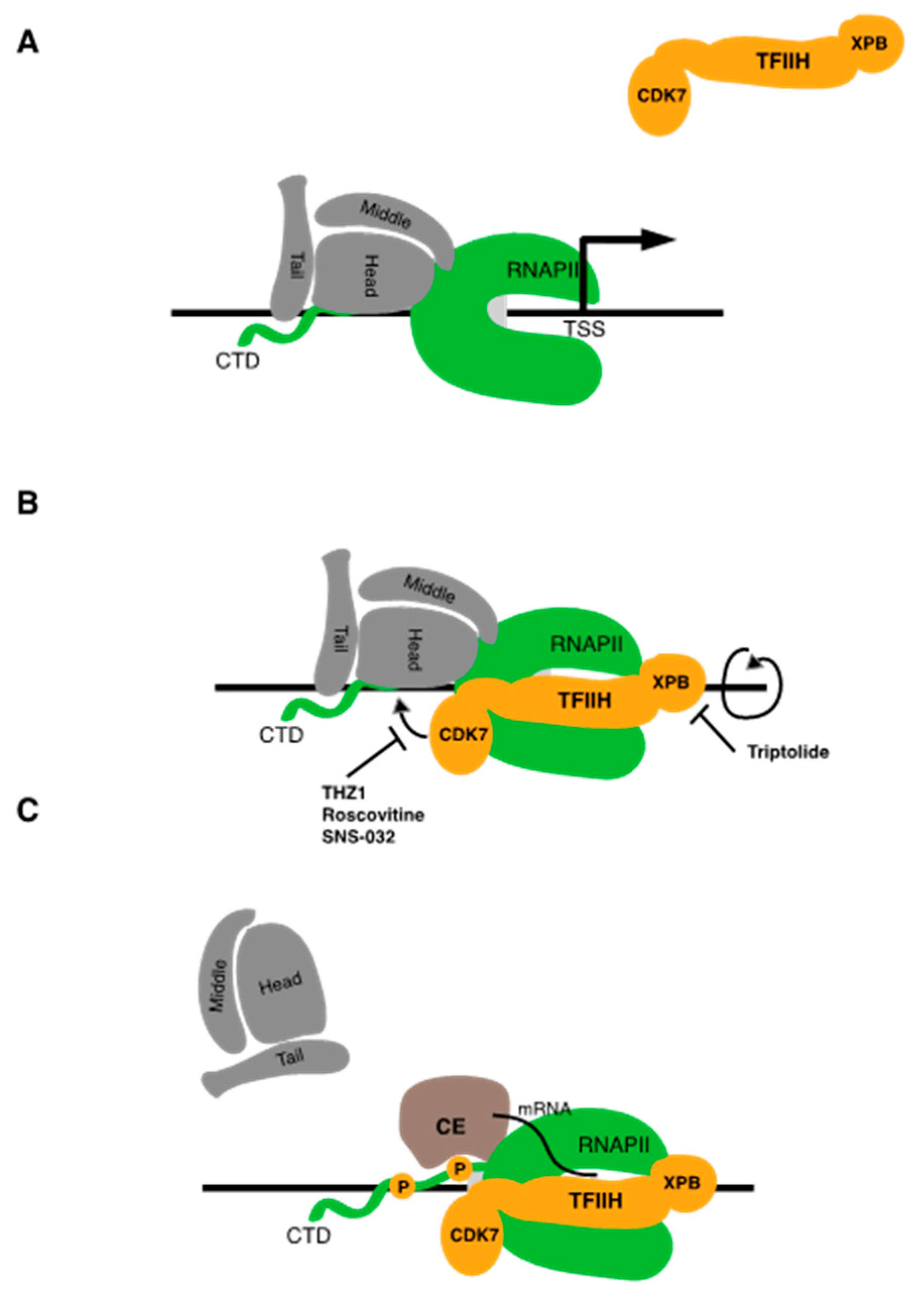
4.2.1. THZ1
4.2.2. Non-Covalent CDK7 Inhibitors
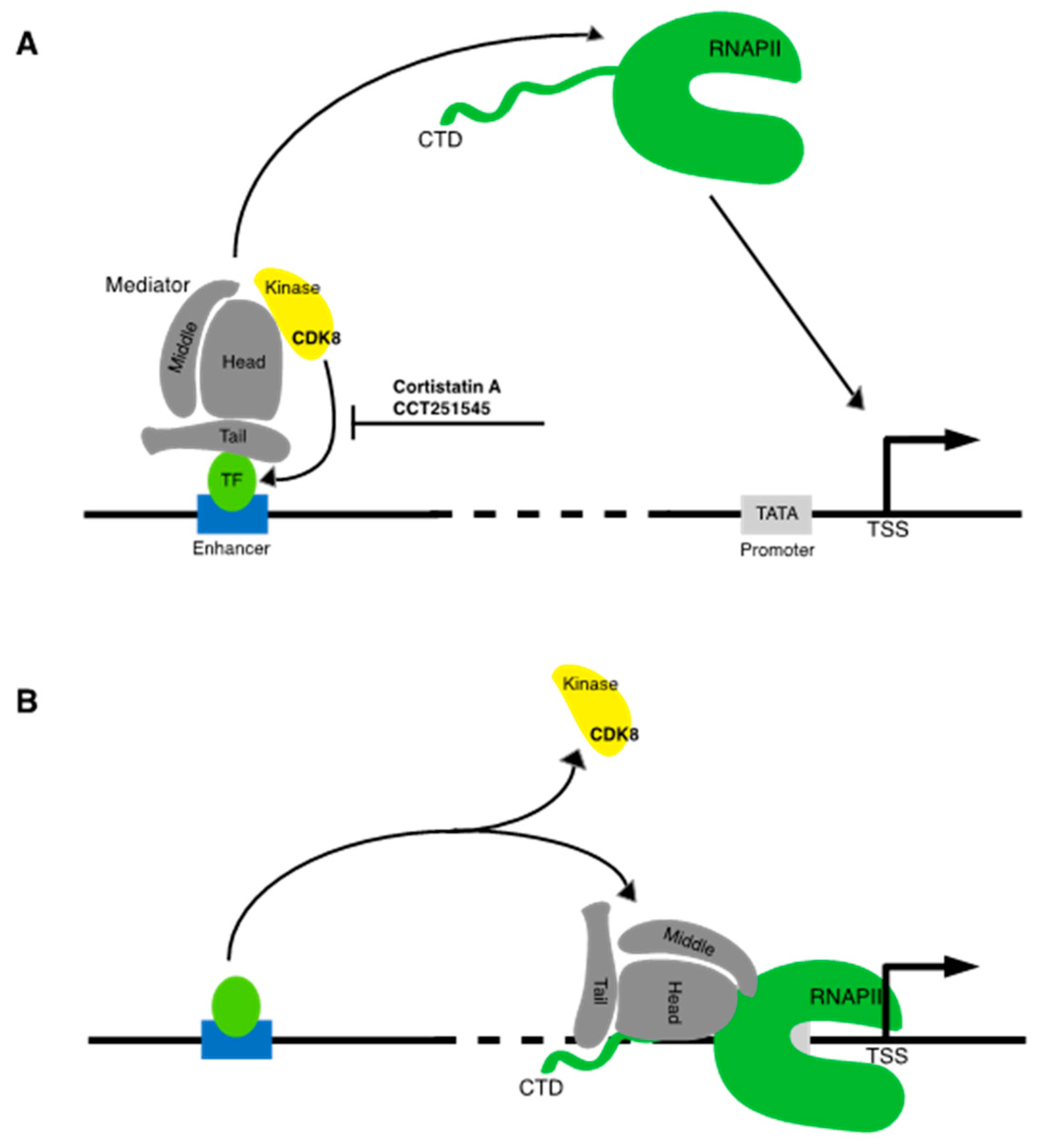
4.2.3. Cortistatin A
4.2.4. Other Mediator Kinase Inhibitors
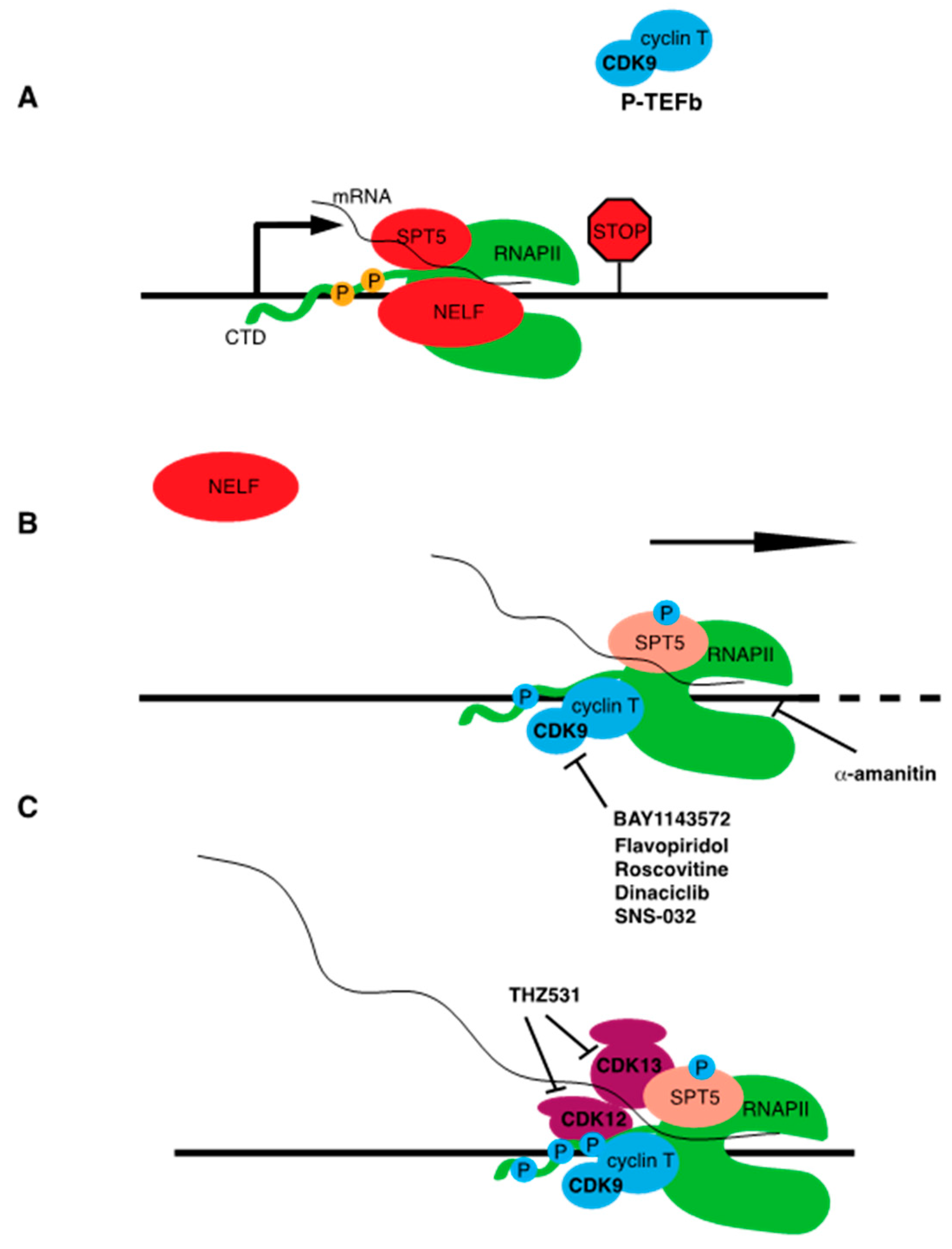
4.2.5. CDK9 Inhibitors
4.2.6. THZ531
5. Triptolide Targets TFIIH Subunit XPB
6. Compounds Targeting RNAPII
7. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Wouters, J.; Kalender Atak, Z.; Aerts, S. Decoding transcriptional states in cancer. Curr. Opin. Genet. Dev. 2017, 43, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Lazo, J.S.; Sharlow, E.R. Drugging Undruggable Molecular Cancer Targets. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 23–40. [Google Scholar] [CrossRef]
- Mapp, A.K.; Pricer, R.; Sturlis, S. Targeting transcription is no longer a quixotic quest. Nat. Chem. Biol. 2015, 11, 891–894. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Chiang, C.M. The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef]
- Warfield, L.; Ramachandran, S.; Baptista, T.; Devys, D.; Tora, L.; Hahn, S. Transcription of Nearly All Yeast RNA Polymerase II-Transcribed Genes Is Dependent on Transcription Factor TFIID. Mol. Cell 2017, 68, 118–129.e5. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Ansari, S.A.; Morse, R.H. Mechanisms of Mediator complex action in transcriptional activation. Cell. Mol. Life Sci. 2013, 70, 2743–2756. [Google Scholar] [CrossRef]
- Jeronimo, C.; Robert, F. The Mediator Complex: At the Nexus of RNA Polymerase II Transcription. Trends Cell Biol. 2017, 27, 765–783. [Google Scholar] [CrossRef]
- Tsai, K.L.; Sato, S.; Tomomori-Sato, C.; Conaway, R.C.; Conaway, J.W.; Asturias, F.J. A conserved Mediator-CDK8 kinase module association regulates Mediator-RNA polymerase II interaction. Nat. Struct. Mol. Biol. 2013, 20, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Jeronimo, C.; Langelier, M.F.; Bataille, A.R.; Pascal, J.M.; Pugh, B.F.; Robert, F. Tail and Kinase Modules Differently Regulate Core Mediator Recruitment and Function In Vivo. Mol. Cell 2016, 64, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, N.; Jin, Y.; Wong, K.H.; Struhl, K. Mediator Undergoes a Compositional Change during Transcriptional Activation. Mol. Cell 2016, 64, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.J.; Trnka, M.J.; Bushnell, D.A.; Davis, R.E.; Mattei, P.J.; Burlingame, A.L.; Kornberg, R.D. Structure of a Complete Mediator-RNA Polymerase II PreInitiation Complex. Cell 2016, 166, 1411–1422.e16. [Google Scholar] [CrossRef] [Green Version]
- Hsin, J.P.; Manley, J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef] [Green Version]
- Fishburn, J.; Tomko, E.; Galburt, E.; Hahn, S. Double-stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3961–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Shuman, S.; Lima, C.D. Structural insights to how mammalian capping enzyme reads the CTD code. Mol. Cell 2011, 43, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, B.; Zeitlinger, J. RNA polymerase II pausing during development. Development 2014, 141, 1179–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missra, A.; Gilmour, D.S. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc. Natl. Acad. Sci. USA 2010, 107, 11301–11306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef]
- McSwiggen, D.T.; Mir, M.; Darzacq, X.; Tjian, R. Evaluating phase separation in live cells: Diagnosis, caveats, and functional consequences. Genes Dev. 2019, 33, 1619–1634. [Google Scholar] [CrossRef]
- Guo, Y.E.; Manteiga, J.C.; Henninger, J.E.; Sabari, B.R.; Dall’Agnese, A.; Hannett, N.M.; Spille, J.H.; Afeyan, L.K.; Zamudio, A.V.; Shrinivas, K.; et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 2019, 572, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Kohoutek, J.; Blazek, D. Cyclin K goes with Cdk12 and Cdk13. Cell Div. 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell. Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paculova, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Rucobo, F.W.; Cramer, P. Structural basis of transcription elongation. Biochim. Biophys. Acta 2013, 1829, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Bushnell, D.A.; Westover, K.D.; Kaplan, C.D.; Kornberg, R.D. Structural basis of transcription: Role of the trigger loop in substrate specificity and catalysis. Cell 2006, 127, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Cramer, P.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 2001, 292, 1863–1876. [Google Scholar] [CrossRef] [Green Version]
- Brueckner, F.; Cramer, P. Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat. Struct. Mol. Biol. 2008, 15, 811–818. [Google Scholar] [CrossRef]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.C.; Novotny-Diermayr, V.; Hart, S.; Ong, L.C.; Loh, Y.K.; Cheong, A.; Tan, Y.C.; Hu, C.; Jayaraman, R.; William, A.D.; et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent antileukemic properties. Leukemia 2012, 26, 236–243. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Kaur, G.; Stetler-Stevenson, M.; Sebers, S.; Worland, P.; Sedlacek, H.; Myers, C.; Czech, J.; Naik, R.; Sausville, E. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J. Natl. Cancer Inst. 1992, 84, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Kaufmann, S.H. Flavopiridol: A cytotoxic flavone that induces cell death in noncycling A549 human lung carcinoma cells. Cancer Res. 1996, 56, 4856–4861. [Google Scholar] [PubMed]
- Kouroukis, C.T.; Belch, A.; Crump, M.; Eisenhauer, E.; Gascoyne, R.D.; Meyer, R.; Lohmann, R.; Lopez, P.; Powers, J.; Turner, R.; et al. Flavopiridol in untreated or relapsed mantle-cell lymphoma: Results of a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 1740–1745. [Google Scholar] [CrossRef]
- Phelps, M.A.; Lin, T.S.; Johnson, A.J.; Hurh, E.; Rozewski, D.M.; Farley, K.L.; Wu, D.; Blum, K.A.; Fischer, B.; Mitchell, S.M.; et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009, 113, 2637–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleyard, M.V.; O’Neill, M.A.; Murray, K.E.; Paulin, F.E.; Bray, S.E.; Kernohan, N.M.; Levison, D.A.; Lane, D.P.; Thompson, A.M. Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model. Int. J. Cancer 2009, 124, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.N.; Xiao, H.Y.; Kim, K.S.; Lu, S.; Han, W.C.; Barbosa, S.A.; Hunt, J.T.; Rawlins, D.B.; Shan, W.; Ahmed, S.Z.; et al. N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J. Med. Chem. 2004, 47, 1719–1728. [Google Scholar] [CrossRef]
- Xie, G.; Tang, H.; Wu, S.; Chen, J.; Liu, J.; Liao, C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int. J. Oncol. 2014, 45, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.L.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Nitulescu, I.I.; Meyer, S.C.; Wen, Q.J.; Crispino, J.D.; Lemieux, M.E.; Levine, R.L.; Pelish, H.E.; Shair, M.D. Mediator Kinase Phosphorylation of STAT1 S727 Promotes Growth of Neoplasms With JAK-STAT Activation. EBioMedicine 2017, 26, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.A.; Ortiz-Ruiz, M.J.; TePoele, R.; Adeniji-Popoola, O.; Box, G.; Court, W.; Czasch, S.; El Bawab, S.; Esdar, C.; Ewan, K.; et al. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. Elife 2016, 5, e20722. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Shi, S.; Huang, S.; Hole, A.J.; Abbas, A.Y.; Baumli, S.; Liu, X.; Lam, F.; Foley, D.W.; Fischer, P.M.; et al. Substituted 4-(thiazol-5-yl)-2-(phenylamino)pyrimidines are highly active CDK9 inhibitors: Synthesis, X-ray crystal structures, structure-activity relationship, and anticancer activities. J. Med. Chem. 2013, 56, 640–659. [Google Scholar] [CrossRef]
- Wang, H.K. The therapeutic potential of flavonoids. Expert Opin. Investig. Drugs 2000, 9, 2103–2119. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M.; Headlee, D.; Stinson, S.F.; Lush, R.M.; Kalil, N.; Villalba, L.; Hill, K.; Steinberg, S.M.; Figg, W.D.; Tompkins, A.; et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J. Clin. Oncol. 1998, 16, 2986–2999. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.S.; Sack, J.S.; Tokarski, J.S.; Qian, L.; Chao, S.T.; Leith, L.; Kelly, Y.F.; Misra, R.N.; Hunt, J.T.; Kimball, S.D.; et al. Thio- and oxoflavopiridols, cyclin-dependent kinase 1-selective inhibitors: Synthesis and biological effects. J. Med. Chem. 2000, 43, 4126–4134. [Google Scholar] [CrossRef]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef]
- Lu, H.; Chang, D.J.; Baratte, B.; Meijer, L.; Schulze-Gahmen, U. Crystal structure of a human cyclin-dependent kinase 6 complex with a flavonol inhibitor, fisetin. J. Med. Chem. 2005, 48, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Carlson, B.A.; Dubay, M.M.; Sausville, E.A.; Brizuela, L.; Worland, P.J. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996, 56, 2973–2978. [Google Scholar]
- Konig, A.; Schwartz, G.K.; Mohammad, R.M.; Al-Katib, A.; Gabrilove, J.L. The novel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2 and induces growth arrest and apoptosis in chronic B-cell leukemia lines. Blood 1997, 90, 4307–4312. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.; Lahusen, T.; Singh, S.; Loaiza-Perez, A.; Worland, P.J.; Pestell, R.; Albanese, C.; Sausville, E.A.; Senderowicz, A.M. Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res. 1999, 59, 4634–4641. [Google Scholar] [PubMed]
- Chao, S.H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, S.H.; Price, D.H. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001, 276, 31793–31799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, L.T.; Pickeral, O.K.; Peng, A.C.; Rosenwald, A.; Hurt, E.M.; Giltnane, J.M.; Averett, L.M.; Zhao, H.; Davis, R.E.; Sathyamoorthy, M.; et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anticancer drug flavopiridol. Genome Biol. 2001, 2, RESEARCH0041. [Google Scholar] [CrossRef] [Green Version]
- Aklilu, M.; Kindler, H.L.; Donehower, R.C.; Mani, S.; Vokes, E.E. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann. Oncol. 2003, 14, 1270–1273. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Ilson, D.; Saltz, L.; O’Reilly, E.; Tong, W.; Maslak, P.; Werner, J.; Perkins, P.; Stoltz, M.; Kelsen, D. Phase II study of the cyclin-dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J. Clin. Oncol. 2001, 19, 1985–1992. [Google Scholar] [CrossRef]
- Stadler, W.M.; Vogelzang, N.J.; Amato, R.; Sosman, J.; Taber, D.; Liebowitz, D.; Vokes, E.E. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in metastatic renal cancer: A University of Chicago Phase II Consortium study. J. Clin. Oncol. 2000, 18, 371–375. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Supko, J.G.; Patterson, A.; Lynch, C.; Lucca, J.; Zacarola, P.F.; Muzikansky, A.; Wright, J.J.; Lynch, T.J., Jr.; Rollins, B.J. A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin. Cancer Res. 2001, 7, 1590–1599. [Google Scholar]
- Lin, T.S.; Howard, O.M.; Neuberg, D.S.; Kim, H.H.; Shipp, M.A. Seventy-two hour continuous infusion flavopiridol in relapsed and refractory mantle cell lymphoma. Leuk. Lymphoma 2002, 43, 793–797. [Google Scholar] [CrossRef]
- Byrd, J.C.; Lin, T.S.; Dalton, J.T.; Wu, D.; Phelps, M.A.; Fischer, B.; Moran, M.; Blum, K.A.; Rovin, B.; Brooker-McEldowney, M.; et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood 2007, 109, 399–404. [Google Scholar] [CrossRef]
- Lin, T.S.; Ruppert, A.S.; Johnson, A.J.; Fischer, B.; Heerema, N.A.; Andritsos, L.A.; Blum, K.A.; Flynn, J.M.; Jones, J.A.; Hu, W.; et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009, 27, 6012–6018. [Google Scholar] [CrossRef]
- Lanasa, M.C.; Andritsos, L.; Brown, J.R.; Gabrilove, J.; Caligaris-Cappio, F.; Ghia, P.; Larson, R.A.; Kipps, T.J.; Leblond, V.; Milligan, D.W.; et al. Final results of EFC6663: A multicenter, international, phase 2 study of alvocidib for patients with fludarabine-refractory chronic lymphocytic leukemia. Leuk. Res. 2015, 39, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Schang, L.M.; Bantly, A.; Knockaert, M.; Shaheen, F.; Meijer, L.; Malim, M.H.; Gray, N.S.; Schaffer, P.A. Pharmacological cyclin-dependent kinase inhibitors inhibit replication of wild-type and drug-resistant strains of herpes simplex virus and human immunodeficiency virus type 1 by targeting cellular, not viral, proteins. J. Virol. 2002, 76, 7874–7882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClue, S.J.; Blake, D.; Clarke, R.; Cowan, A.; Cummings, L.; Fischer, P.M.; MacKenzie, M.; Melville, J.; Stewart, K.; Wang, S.; et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int. J. Cancer 2002, 102, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Coker-Gurkan, A.; Arisan, E.D.; Obakan, P.; Ozfiliz, P.; Kose, B.; Bickici, G.; Palavan-Unsal, N. Roscovitine-treated HeLa cells finalize autophagy later than apoptosis by downregulating Bcl2. Mol. Med. Rep. 2015, 11, 1968–1974. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Kim, S.U.; Shin, D.Y.; Choi, K.S. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene 2004, 23, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Garrofe-Ochoa, X.; Cosialls, A.M.; Ribas, J.; Gil, J.; Boix, J. Transcriptional modulation of apoptosis regulators by roscovitine and related compounds. Apoptosis 2011, 16, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Raynaud, F.I.; Whittaker, S.R.; Fischer, P.M.; McClue, S.; Walton, M.I.; Barrie, S.E.; Garrett, M.D.; Rogers, P.; Clarke, S.J.; Kelland, L.R.; et al. In vitro and in vivo pharmacokinetic-pharmacodynamic relationships for the trisubstituted aminopurine cyclin-dependent kinase inhibitors olomoucine, bohemine and CYC202. Clin. Cancer Res. 2005, 11, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [PubMed]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, A.; Stockett, D.E.; Walker, D.; Arkin, M.R.; Hoch, U.; Fox, J.A.; Hawtin, R.E. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother. Pharmacol. 2009, 64, 723–732. [Google Scholar] [CrossRef]
- Walsby, E.; Lazenby, M.; Pepper, C.; Burnett, A.K. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine. Leukemia 2011, 25, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Loschmann, N.; Michaelis, M.; Rothweiler, F.; Zehner, R.; Cinatl, J.; Voges, Y.; Sharifi, M.; Riecken, K.; Meyer, J.; von Deimling, A.; et al. Testing of SNS-032 in a Panel of Human Neuroblastoma Cell Lines with Acquired Resistance to a Broad Range of Drugs. Transl. Oncol. 2013, 6, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of Dinaciclib (SCH 727965): A Potent and Selective Inhibitor of Cyclin-Dependent Kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.F.; Lin, J.D.; Hsueh, C.; Chou, T.C.; Wong, R.J. A cyclin-dependent kinase inhibitor, dinaciclib in preclinical treatment models of thyroid cancer. PLoS ONE 2017, 12, e0172315. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Ghia, P.; Scarfo, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ni Chonghaile, T.; Fan, Y.; Madden, S.F.; Klinger, R.; O’Connor, A.E.; Walsh, L.; O’Hurley, G.; Mallya Udupi, G.; Joseph, J.; et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3834–3845. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Ali, S.; Heathcote, D.A.; Kroll, S.H.; Jogalekar, A.S.; Scheiper, B.; Patel, H.; Brackow, J.; Siwicka, A.; Fuchter, M.J.; Periyasamy, M.; et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009, 69, 6208–6215. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Periyasamy, M.; Sava, G.P.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; et al. ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol. Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Cortistatins, A, B, C, and D; antiangiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J. Am. Chem. Soc. 2006, 128, 3148–3149. [Google Scholar] [CrossRef]
- Aoki, S.; Watanabe, Y.; Tanabe, D.; Arai, M.; Suna, H.; Miyamoto, K.; Tsujibo, H.; Tsujikawa, K.; Yamamoto, H.; Kobayashi, M. Structure-activity relationship and biological property of cortistatins, antiangiogenic spongean steroidal alkaloids. Bioorg. Med. Chem. 2007, 15, 6758–6762. [Google Scholar] [CrossRef]
- Cee, V.J.; Chen, D.Y.; Lee, M.R.; Nicolaou, K.C. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew. Chem. Int. Ed. Engl. 2009, 48, 8952–8957. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Dale, T.; Clarke, P.A.; Esdar, C.; Waalboer, D.; Adeniji-Popoola, O.; Ortiz-Ruiz, M.J.; Mallinger, A.; Samant, R.S.; Czodrowski, P.; Musil, D.; et al. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. [Google Scholar] [CrossRef]
- Rzymski, T.; Mikula, M.; Zylkiewicz, E.; Dreas, A.; Wiklik, K.; Golas, A.; Wojcik, K.; Masiejczyk, M.; Wrobel, A.; Dolata, I.; et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017, 8, 33779–33795. [Google Scholar] [CrossRef] [Green Version]
- Baumli, S.; Endicott, J.A.; Johnson, L.N. Halogen bonds form the basis for selective P-TEFb inhibition by DRB. Chem. Biol. 2010, 17, 931–936. [Google Scholar] [CrossRef] [Green Version]
- Hole, A.J.; Baumli, S.; Shao, H.; Shi, S.; Huang, S.; Pepper, C.; Fischer, P.M.; Wang, S.; Endicott, J.A.; Noble, M.E. Comparative structural and functional studies of 4-(thiazol-5-yl)-2-(phenylamino)pyrimidine-5-carbonitrile CDK9 inhibitors suggest the basis for isotype selectivity. J. Med. Chem. 2013, 56, 660–670. [Google Scholar] [CrossRef]
- Lu, H.; Xue, Y.; Yu, G.K.; Arias, C.; Lin, J.; Fong, S.; Faure, M.; Weisburd, B.; Ji, X.; Mercier, A.; et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife 2015, 4, e06535. [Google Scholar] [CrossRef]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- Lucking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12, 1776–1793. [Google Scholar] [CrossRef]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 36. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.R.; Dai, Y.; Zhao, J.; Lin, L.; Wang, Y.; Wang, Y. A Mechanistic Overview of Triptolide and Celastrol, Natural Products from Tripterygium wilfordii Hook F. Front. Pharmacol. 2018, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Titov, D.V.; Gilman, B.; He, Q.L.; Bhat, S.; Low, W.K.; Dang, Y.; Smeaton, M.; Demain, A.L.; Miller, P.S.; Kugel, J.F.; et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011, 7, 182–188. [Google Scholar] [CrossRef] [Green Version]
- He, Q.L.; Minn, I.; Wang, Q.; Xu, P.; Head, S.A.; Datan, E.; Yu, B.; Pomper, M.G.; Liu, J.O. Targeted Delivery and Sustained Antitumor Activity of Triptolide through Glucose Conjugation. Angew. Chem. Int. Ed. Engl. 2016, 55, 12035–12039. [Google Scholar] [CrossRef]
- Bensaude, O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2011, 2, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Musso, L.; Mazzini, S.; Rossini, A.; Castagnoli, L.; Scaglioni, L.; Artali, R.; Di Nicola, M.; Zunino, F.; Dallavalle, S. c-MYC G-quadruplex binding by the RNA polymerase I inhibitor BMH-21 and analogues revealed by a combined NMR and biochemical Approach. Biochim. Biophys. Acta 2018, 1862, 615–629. [Google Scholar] [CrossRef]
- Kaplan, C.D.; Larsson, K.M.; Kornberg, R.D. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Mol. Cell 2008, 30, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Kume, K.; Ikeda, M.; Miura, S.; Ito, K.; Sato, K.A.; Ohmori, Y.; Endo, F.; Katagiri, H.; Ishida, K.; Ito, C.; et al. alpha-Amanitin Restrains Cancer Relapse from Drug-Tolerant Cell Subpopulations via TAF15. Sci. Rep. 2016, 6, 25895. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, X.; Han, C.; Wan, G.; Huang, X.; Ivan, C.; Jiang, D.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Rao, P.H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701. [Google Scholar] [CrossRef] [Green Version]
- Clark, V.E.; Harmanci, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Pan-CDK Inhibitors | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| Flavopiridol (Alvocidib) | CDK1 - IC50 30 nM [41] CDK2 - IC50 170 nM [42] CDK4 - IC50 100 nM [41] CDK7 - IC50 875 nM [42] CDK9 - IC50 20 nM [42] |  | Three phase I trials ongoing for acute myeloid leukemia in combination with venetoclax and decitabine |
| Roscovitine (Seleciclib) | CDK1 - IC50 650 nM [43] CDK2 - IC50 700 nM [43] CDK5 - IC50 160 nM [43] CDK7 - IC50 460 nM [43] CDK9 - IC50 600 nM [44] |  | Single-agent phase II trial for Cushing’s disease |
| SNS-032 | CDK2 - IC50 48 nM [45] CDK7 - IC50 62 nM [45] CDK9 - IC50 4 nM [45] |  | None currently ongoing |
| Dinaciclib | CDK1 - IC50 3 nM [46] CDK2 - IC50 1 nM [46] CDK5 - IC50 5 nM [46] CDK9 - IC50 9 nM [46] |  | Four phase I combination (venetoclax, pembrolizumab, veliparib) and single-agent trials and one phase II trial for melanoma |
| AT7519M | CDK1 - IC50 210 nM [38] CDK2 - IC50 47 nM [38] CDK4 - IC50 100 nM [38] CDK5 - IC50 13 nM [38] CDK6 - IC50 170 nM [38] CDK9 - IC50 <10 nM [38] |  | One phase I trial in combination with onalespib for solid tumors |
| TG02 | CDK1 - IC50 9 nM [39] CDK2 - IC50 5 nM [39] CDK3 - IC50 8 nM [39] CDK5 - IC50 4 nM [39] CDK9 - IC50 3 nM [39] |  | Three phase I studies for glioblastoma in combination with temozolomide (TMZ), and one for glioma in combination with pembrolizumab and TMZ |
| CDK7 | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| THZ1 | CDK7 - IC50 3.2 nM [47] CDK12 - IC50 250 nM [47] |  | None currently ongoing |
| SY-1365 | CDK2 - IC50 2600 nM CDK7 - IC50 20 nM CDK9 - IC50 670 nM |  | Single-agent phase I for advanced solid tumors |
| BS-181 | CDK2 - IC50 880 nM [48] CDK7 - IC50 20 nM [48] others - >1000 nM [48] |  | None currently ongoing |
| ICEC0942 (CT7001) | CDK2 - IC50 620 nM [49] CDK7 - IC50 4 nM [49] CDK9 - IC50 1200 nM [49] others - >1000 nM [49] |  | Phase I/II for advanced malignancies |
| Mediator Kinase | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| Cortistatin A | Cdk8 - IC50 12 nM Cdk8 - Kd 17 nM Cdk19 - Kd 10 nM |  | None currently ongoing |
| CCT251545 | CDK8 - IC50 5 nM [48] CDK19 - IC50 6 nM [48] |  | None currently ongoing |
| SEL120-34A | CDK8 - IC50 4.4 nM [49] CDK19 - IC50 10.4 nM [49] |  | None currently ongoing |
| CDK9 | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| BAY1143572 (Atuveciclib) | CDK9 - IC50 13 nM [50] |  | None currently ongoing |
| BAY1251152 | CDK9 - IC50 3 nM |  | Single-agent phase I trial for advanced solid neoplasms |
| CDK12 and CDK13 | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| THZ531 | CDK12 - IC50 158 nM [29] CDK13 - IC50 69 nM [29] |  | None currently ongoing |
| TFIIH subunit XPB | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| Triptolide | XPB ATPase - IC50 145 nM [51] |  | Phase I trials for advanced solid tumors in combination with paclitaxel and for HIV; phase II trial for refractory pancreatic cancer |
| RNA Polymerase | |||
| Inhibitor Name | Target Specificity | Compound Structure | Listed Clinical Trials |
| Actinomycin D | RNAPI > RNAPII > RNAPIII [52] |  | Four phase II and six phase III trials as single-agent or combination therapies for various cancers |
| α-amanitin | RNAPII [52] |  | None currently ongoing |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, R.D.; Hébert, T.E.; Tanny, J.C. Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. Int. J. Mol. Sci. 2020, 21, 3354. https://doi.org/10.3390/ijms21093354
Martin RD, Hébert TE, Tanny JC. Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. International Journal of Molecular Sciences. 2020; 21(9):3354. https://doi.org/10.3390/ijms21093354
Chicago/Turabian StyleMartin, Ryan D., Terence E. Hébert, and Jason C. Tanny. 2020. "Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery" International Journal of Molecular Sciences 21, no. 9: 3354. https://doi.org/10.3390/ijms21093354
APA StyleMartin, R. D., Hébert, T. E., & Tanny, J. C. (2020). Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. International Journal of Molecular Sciences, 21(9), 3354. https://doi.org/10.3390/ijms21093354