Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction

 and
and
Abstract
:1. Introduction
2. Results
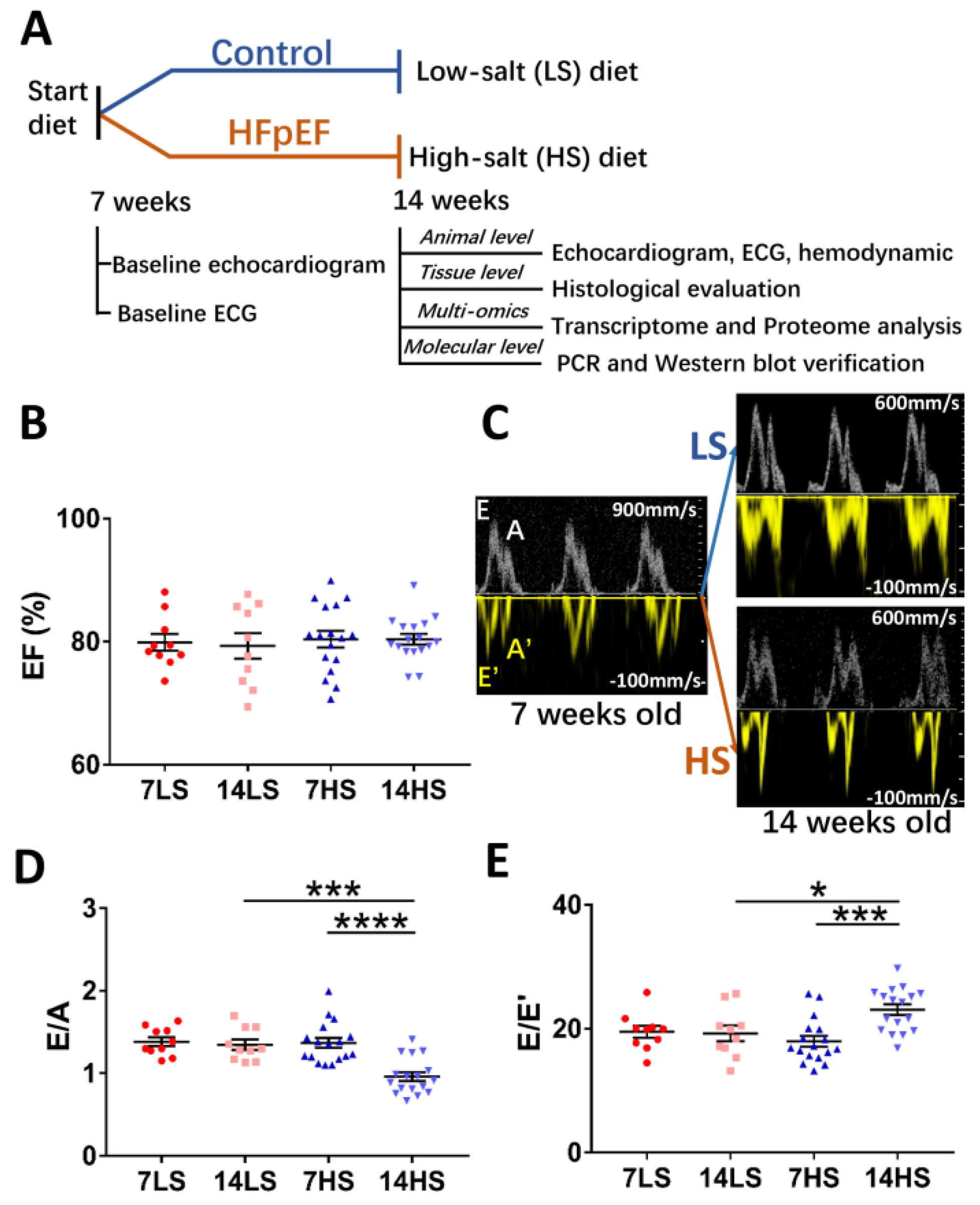
2.1. Development of HFpEF in HS-Fed Rats
2.2. ECG Analyses in Control and HFpEF Rats
2.3. Hemodynamic Analysis
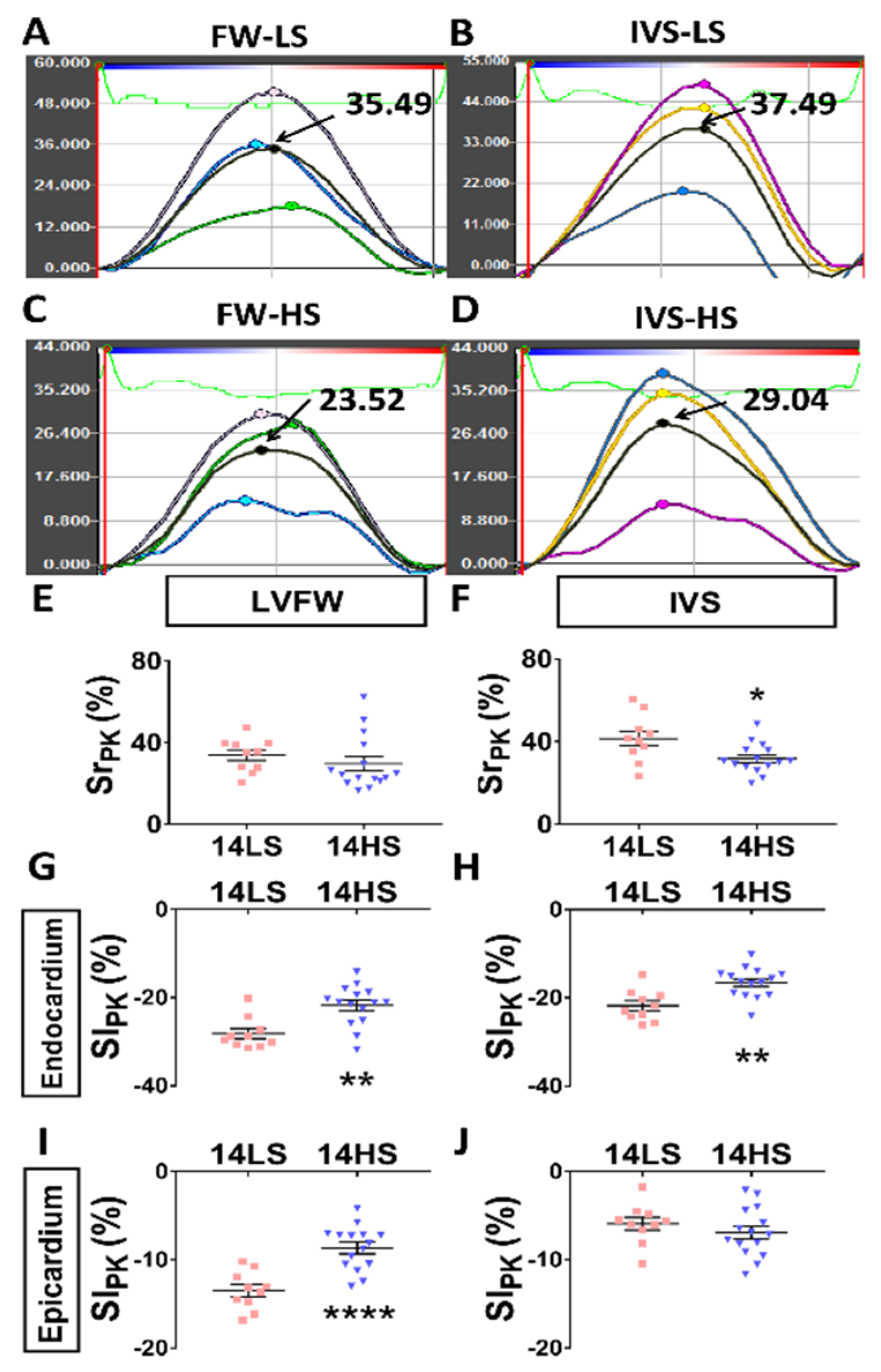
2.4. Histological Evaluation
2.5. Proteomic Study and Western Blot
2.6. Microarray Study and Quantitative Polymerase Chain Reaction
3. Discussion
3.1. Pathophysiological Changes in Organ and Tissue Levels
3.2. Promising Regulatory Gene Targets for Myocardial and Myocyte Stiffening
3.3. Promising Regulatory Gene Targets for Myocyte Apoptosis
3.4. Critique of the Study
4. Materials and Methods
4.1. Study Design
4.2. Echocardiographic Measurements
4.3. Surface Electrocardiogram
4.4. Speckle Tracking Echocardiography (STE) Measurements
4.5. Doppler-Mode Tracings
4.6. Hemodynamic Measurements
4.7. Histological Evaluation
4.8. Shotgun Proteomics Analysis
4.9. Microarray Analysis
4.10. Bioinformatics
4.11. Western Blot
4.12. Quantitative RT-PCR
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| EF | Ejection fraction |
| LV | Left ventricular |
| DSS | Dahl salt-sensitive |
| HS | High-salt |
| LS | Low-salt |
| GSEA | Gene Set Enrichment Analysis |
| GO | Gene Ontology |
References
- Safar, M.E. Arterial stiffness as a risk factor for clinical hypertension. Nat. Rev. Cardiol. 2018, 15, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.; Butlin, M.; Liu, Y.Y.; Ng, K.; Avolio, A.P. Heart Rate Dependence of Aortic Pulse Wave Velocity at Different Arterial Pressures in Rats. Hypertension 2012, 60, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zile, M.R.; Baicu, C.F.; Gaasch, W.H. Diastolic heart failure--abnormalities in active relaxation and passive stiffness of the left ventricle. N. Engl. J. Med. 2004, 350, 1953–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.H.; Kim, Y.H.; Roh, J.H.; Kang, J.W.; Ahn, J.M.; Kweon, J.; Lee, J.B.; Choi, S.H.; Shin, E.S.; Park, D.W.; et al. Diagnostic performance of on-site CT-derived fractional flow reserve versus CT perfusion. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Brucks, S.; Little, W.C.; Chao, T.; Kitzman, D.W. Contribution of left ventricular diastolic dysfunction to heart failure regardless of ejection fraction. Am. J. Cardiol. 2005, 95, 603–606. [Google Scholar] [CrossRef]
- Taylor, M.G. An approach to an analysis of the arterial pulse wave. II. Fluid oscillations in an elastic pipe. Phys. Med. Biol. 1957, 1, 321–329. [Google Scholar] [CrossRef]
- American College of Cardiology Foundation Task Force on Expert Consensus, D.; Mark, D.B.; Berman, D.S.; Budoff, M.J.; Carr, J.J.; Gerber, T.C.; Hecht, H.S.; Hlatky, M.A.; Hodgson, J.M.; Lauer, M.S.; et al. ACCF/ACR/AHA/NASCI/SAIP/SCAI/SCCT 2010 expert consensus document on coronary computed tomographic angiography: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. Circulation 2010, 121, 2509–2543. [Google Scholar] [CrossRef]
- American College of Cardiology Foundation Task Force on Expert Consensus, D.; Mark, D.B.; Berman, D.S.; Budoff, M.J.; Carr, J.J.; Gerber, T.C.; Hecht, H.S.; Hlatky, M.A.; Hodgson, J.M.; Lauer, M.S.; et al. ACCF/ACR/AHA/NASCI/SAIP/SCAI/SCCT 2010 expert consensus document on coronary computed tomographic angiography: A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. J. Am. Coll. Cardiol. 2010, 55, 2663–2699. [Google Scholar] [CrossRef]
- Gomes, A.C.; Falcao-Pires, I.; Pires, A.L.; Bras-Silva, C.; Leite-Moreira, A.F. Rodent models of heart failure: An updated review. Heart Fail. Rev. 2013, 18, 219–249. [Google Scholar] [CrossRef] [Green Version]
- Klotz, S.; Hay, I.; Zhang, G.; Maurer, M.; Wang, J.; Burkhoff, D. Development of heart failure in chronic hypertensive Dahl rats: Focus on heart failure with preserved ejection fraction. Hypertension 2006, 47, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Rambihar, S.; Gao, P.; Teo, K.; Bohm, M.; Yusuf, S.; Lonn, E.; Investigators, O.T. Heart Rate is associated with Increased Risk of Major Cardiovascular Events, Cardiovascular and All-Cause Death in Patients with Stable Chronic Cardiovascular Disease—An Analysis of ONTARGET/TRANSCEND. Circulation 2010, 122, A12667. [Google Scholar]
- Taylor, N.E.; Glocka, P.; Liang, M.; Cowley, A.W., Jr. NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl, S. rats. Hypertension 2006, 47, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trolliet, M.R.; Rudd, M.A.; Loscalzo, J. Oxidative stress and renal dysfunction in salt-sensitive hypertension. Kidney Blood Press. Res. 2001, 24, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Nishiyama, A.; Rahman, M.; Nagai, Y.; Noma, T.; Namba, T.; Ishizawa, M.; Murakami, K.; Miyatake, A.; Kimura, S. Contribution of reactive oxygen species to the pathogenesis of left ventricular failure in Dahl salt-sensitive hypertensive rats: Effects of angiotensin II blockade. J. Hypertens. 2006, 24, 1097–1104. [Google Scholar] [CrossRef]
- Tsutsui, H.; Ide, T.; Hayashidani, S.; Kinugawa, S.; Suematsu, N.; Utsumi, H.; Takeshita, A. Effects of ACE inhibition on left ventricular failure and oxidative stress in Dahl salt-sensitive rats. J. Cardiovasc. Pharmacol. 2001, 37, 725–733. [Google Scholar] [CrossRef]
- Bayorh, M.A.; Ganafa, A.A.; Socci, R.R.; Silvestrov, N.; Abukhalaf, I.K. The role of oxidative stress in salt-induced hypertension. Am. J. Hypertens. 2004, 17, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Takatsu, M.; Hattori, T.; Murase, T.; Ohtake, M.; Kato, M.; Nashima, K.; Nakashima, C.; Takahashi, K.; Ito, H.; Niinuma, K. Comparison of the effects of cilnidipine and amlodipine on cardiac remodeling and diastolic dysfunction in Dahl salt-sensitive rats. J. Hypertens. 2012, 30, 1845–1855. [Google Scholar] [CrossRef]
- Huang, P.; Chen, S.; Wang, Y.; Liu, J.; Yao, Q.; Huang, Y.; Li, H.; Zhu, M.; Wang, S.; Li, L. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide 2015, 46, 192–203. [Google Scholar] [CrossRef]
- Huang, B.S.; Van Vliet, B.N.; Leenen, F.H. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1160–H1166. [Google Scholar] [CrossRef]
- Huang, B.S.; Veerasingham, S.J.; Leenen, F.H. Brain “ouabain,” ANG II, and sympathoexcitation by chronic central sodium loading in rats. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H1269–H1276. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Veerasingham, S.J.; Leenen, F.H. Activation of brain renin-angiotensin-aldosterone system by central sodium in Wistar rats. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1109–H1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.S.; White, R.A.; Jeng, A.Y.; Leenen, F.H. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in Dahl salt-sensitive rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, R994–R1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [PubMed]
- Hughes, A.D.; Parker, K.H. Forward and backward waves in the arterial system: Impedance or wave intensity analysis? Med. Biol. Eng. Comput. 2009, 47, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Borbely, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.J.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, V.; Teofilovski-Parapid, G.; Stankovic, G.; Parapid, B.; Malobabic, S.; Stojic, V. Third coronary artery in monkey heart. Acta Vet. Hung. 2004, 52, 253–257. [Google Scholar] [CrossRef]
- Ngo, J.P.; Ow, C.P.; Gardiner, B.S.; Kar, S.; Pearson, J.T.; Smith, D.W.; Evans, R.G. Diffusive shunting of gases and other molecules in the renal vasculature: Physiological and evolutionary significance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R797–R810. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.I.; Spaan, J.A. Pressure-flow relations in coronary circulation. Physiol. Rev. 1990, 70, 331–390. [Google Scholar] [CrossRef]
- Bassingthwaighte, J.B.; Van Beek, J.H.; King, R.B. Fractal branchings: The basis of myocardial flow heterogeneities? Ann. N. Y. Acad. Sci. 1990, 591, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001, 70, 281–312. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Kuang, Y.; Wu, Y.; Xie, W.; Simon, M.I.; Wu, D. Roles of phospholipase C β2 in chemoattractant-elicited responses. Proc. Natl. Acad. Sci. USA 1997, 94, 7971–7975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, M.A.; Maxfield, F.R. Ca2+-and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature 1995, 377, 75. [Google Scholar] [CrossRef] [PubMed]
- Taruno, A.; Vingtdeux, V.; Ohmoto, M.; Ma, Z.; Dvoryanchikov, G.; Li, A.; Adrien, L.; Zhao, H.; Leung, S.; Abernethy, M. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 2013, 495, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Smith, N.P.; Pullan, A.J.; Hunter, P.J. Generation of an anatomically based geometric coronary model. Ann. Biomed. Eng. 2000, 28, 14–25. [Google Scholar] [CrossRef]
- Wendt, N.; Schulz, A.; Qadri, F.; Bolbrinker, J.; Kossmehl, P.; Winkler, K.; Stoll, M.; Vetter, R.; Kreutz, R. Genetic analysis of salt-sensitive hypertension in Dahl rats reveals a link between cardiac fibrosis and high cholesterol. Cardiovasc. Res. 2009, 81, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, E.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Dong, Y.F.; Matsuba, S.; Ogawa, H.; Kim-Mitsuyama, S. Role of xanthine oxidoreductase in the reversal of diastolic heart failure by candesartan in the salt-sensitive hypertensive rat. Hypertension 2007, 50, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyachi, M.; Yazawa, H.; Furukawa, M.; Tsuboi, K.; Ohtake, M.; Nishizawa, T.; Hashimoto, K.; Yokoi, T.; Kojima, T.; Murate, T.; et al. Exercise training alters left ventricular geometry and attenuates heart failure in dahl salt-sensitive hypertensive rats. Hypertension 2009, 53, 701–707. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heerebeek, L.; Paulus, W. Understanding heart failure with preserved ejection fraction: Where are we today? Neth. Heart J. 2016, 24, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, Y.; Weston, S.A.; Redfield, M.M.; Chamberlain, A.M.; Manemann, S.M.; Jiang, R.; Killian, J.M.; Roger, V.L. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 2015, 175, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Souders, C.A.; Borg, T.K.; Banerjee, I.; Baudino, T.A. Pressure overload induces early morphological changes in the heart. Am. J. Pathol. 2012, 181, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.I. Heterogeneity of myocardial blood flow. Basic Res. Cardiol. 1995, 90, 103–111. [Google Scholar] [CrossRef]
- Austin, R.E., Jr.; Aldea, G.S.; Coggins, D.L.; Flynn, A.E.; Hoffman, J.I. Profound spatial heterogeneity of coronary reserve. Discordance between patterns of resting and maximal myocardial blood flow. Circ. Res. 1990, 67, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Ferrucci, A.; Ciavarella, G.M.; De Paolis, P.; Venturelli, V.; Tocci, G.; De Biase, L.; Rubattu, S.; Volpe, M. Markers of inflammation and fibrosis are related to cardiovascular damage in hypertensive patients with metabolic syndrome. Am. J. Hypertens. 2007, 20, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Riad, A.; Dhayat, N.; Spillmann, F.; Du, J.; Dhayat, S.; Westermann, D.; Hilfiker-Kleiner, D.; Noutsias, M.; Laufs, U. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia 2007, 50, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Marin-Garcia, J.; Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016, 21, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Specht, H. Handbook of physiology. Science 1966, 152, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, J.S.; Kumar, S.; Vijayan, M.; Bhatti, G.K.; Reddy, P.H. Therapeutic Strategies for Mitochondrial Dysfunction and Oxidative Stress in Age-Related Metabolic Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 146, 13–46. [Google Scholar] [PubMed]
- Konidala, S.; Gutterman, D.D. Coronary vasospasm and the regulation of coronary blood flow. Prog. Cardiovasc. Dis. 2004, 46, 349–373. [Google Scholar] [CrossRef]
- Adkins, J.E.; Rivlin, R.S. Large Elastic Deformations of Isotropic Materials. 9. The Deformation of Thin Shells. Philos. Trans. R. Soc. Lond. Ser. Math. Phys. Sci. 1952, 244, 505–531. [Google Scholar]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure with Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Wu, H.; Li, L.; Niu, P.; Huang, X.; Liu, J.; Zhang, F.; Shen, W.; Tan, W.; Wu, Y.; Huo, Y. The Structure-function remodeling in rabbit hearts of myocardial infarction. Physiol. Rep. 2017, 5, e13311. [Google Scholar] [CrossRef]
- Sahn, D.J.; DeMaria, A.; Kisslo, J.; Weyman, A. Recommendations regarding quantitation in M-mode echocardiography: Results of a survey of echocardiographic measurements. Circulation 1978, 58, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Zhang, W.; Zhao, D.; Sulejmani, F.; Feng, Y.; Huo, Y.; Tan, W. Cardiac wall mechanics analysis in hypertension-induced heart failure rats with preserved ejection fraction. J. Biomech. 2020, 98, 109428. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Khoury, D.S.; Yue, Y.; Torre-Amione, G.; Nagueh, S.F. Preserved left ventricular twist and circumferential deformation, but depressed longitudinal and radial deformation in patients with diastolic heart failure. Eur. Heart J. 2008, 29, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Kirshenbaum, L.A.; de Moissac, D. The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation 1997, 96, 1580–1585. [Google Scholar] [CrossRef]
- Zhong, L.; Zhou, J.; Chen, X.; Liu, J.; Liu, Z.; Chen, Y.; Bai, Y. Quantitative proteomics reveals EVA1A-related proteins involved in neuronal differentiation. Proteomics 2017, 17, 1600294. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | No. | LVEDD (mm) | LVESD (mm) | LV/BW (g/kg) | Heart/BW (g/kg) | Lung/BW (g/kg) | Kidney/BW (g/kg) |
|---|---|---|---|---|---|---|---|
| 14LS | 8 | 7.10 ± 0.10 | 3.80 ± 0.25 | 2.37 ± 0.10 | 3.60 ± 0.11 | 3.96 ± 0.05 | 3.73 ± 0.16 |
| 14HS | 8 | 7.17 ± 0.23 | 4.11 ± 0.43 | 3.42 ± 0.26 ** | 4.06 ± 0.15 * | 4.18 ± 0.06 * | 4.31 ± 0.08 ** |
| Group | No. | LVEDP (mmHg) | dP/dt max (mmHg/s) | dP/dt min (mmHg/s) | ASP (mmHg) | ADP (mmHg) | MAP (mmHg) |
|---|---|---|---|---|---|---|---|
| 14LS | 8 | 3.72 ± 0.38 | 9895 ± 833 | −6890 ± 528 | 160.5 ± 7.9 | 106.5 ± 7.7 | 124.5 ± 6.9 |
| 14HS | 8 | 11.14 ± 2.03 ** | 7010 ± 941 * | −5051 ± 489 * | 184.6 ± 6.8 * | 128.2 ± 4.4 * | 147.0 ± 4.9 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Zhang, H.; Yao, W.; Li, L.; Niu, P.; Huo, Y.; Tan, W. Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction. Int. J. Mol. Sci. 2020, 21, 3362. https://doi.org/10.3390/ijms21093362
Zhang W, Zhang H, Yao W, Li L, Niu P, Huo Y, Tan W. Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction. International Journal of Molecular Sciences. 2020; 21(9):3362. https://doi.org/10.3390/ijms21093362
Chicago/Turabian StyleZhang, Wenxi, Huan Zhang, Weijuan Yao, Li Li, Pei Niu, Yunlong Huo, and Wenchang Tan. 2020. "Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction" International Journal of Molecular Sciences 21, no. 9: 3362. https://doi.org/10.3390/ijms21093362
APA StyleZhang, W., Zhang, H., Yao, W., Li, L., Niu, P., Huo, Y., & Tan, W. (2020). Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction. International Journal of Molecular Sciences, 21(9), 3362. https://doi.org/10.3390/ijms21093362