Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt–Jakob Disease Patients

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
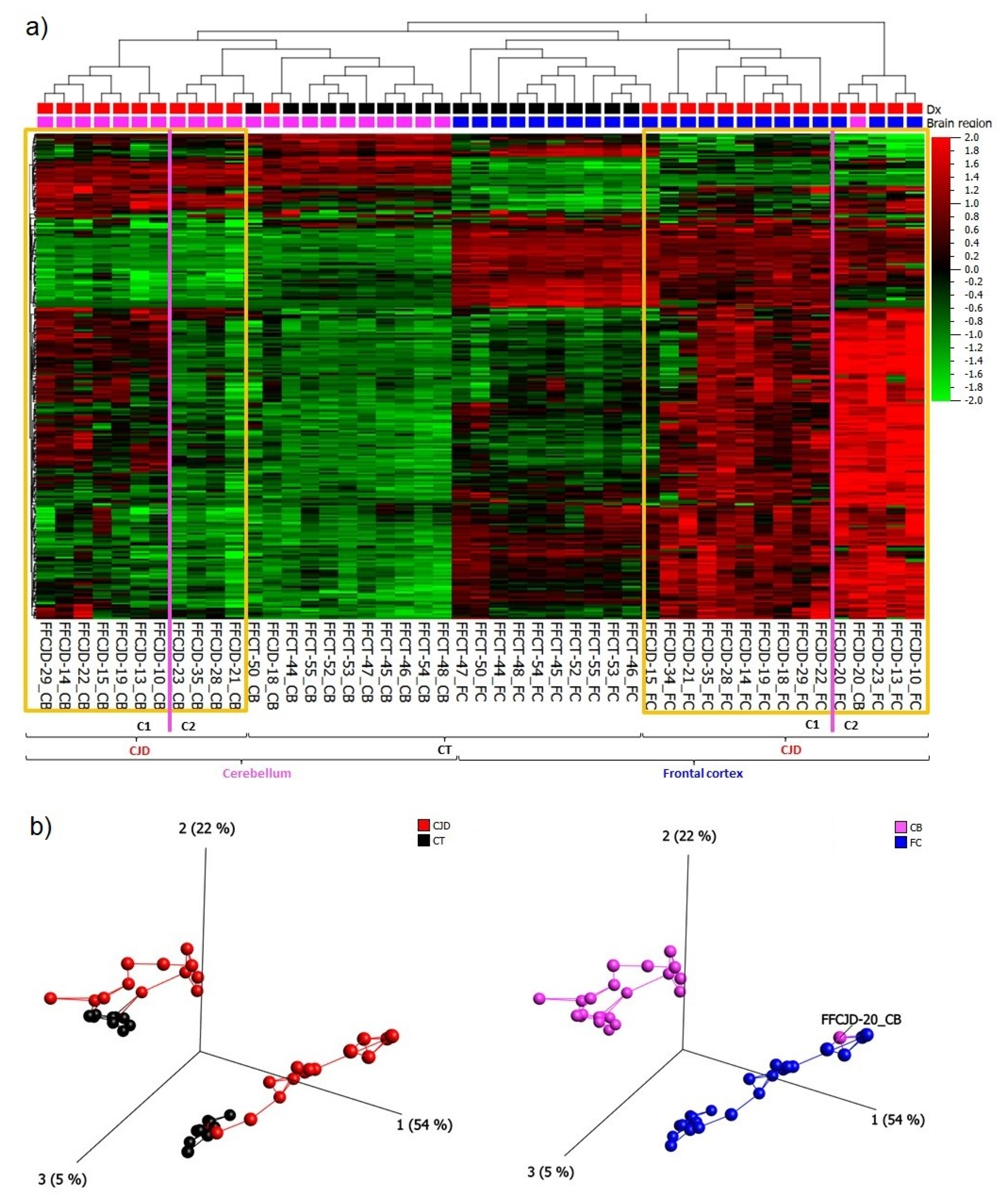
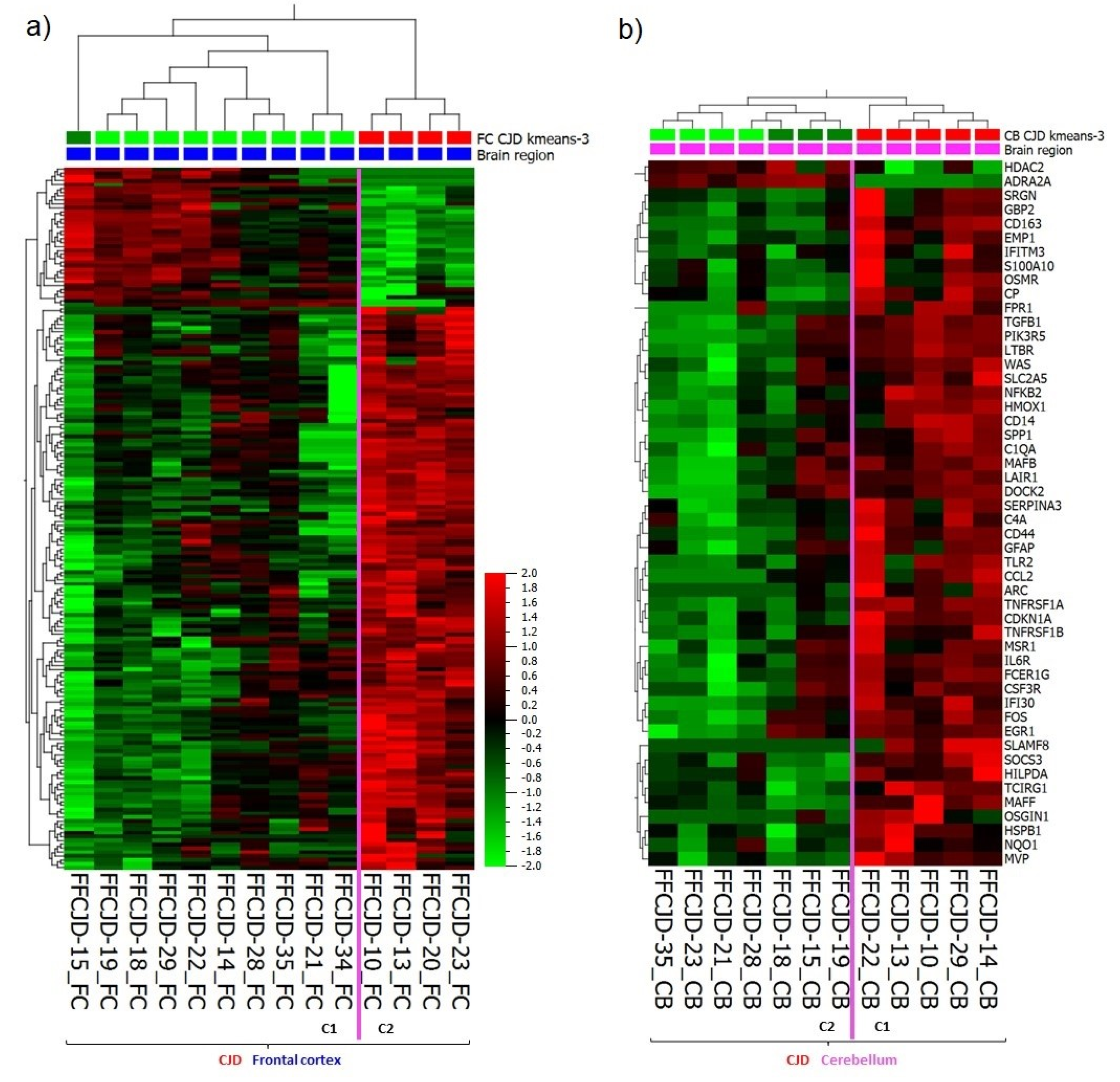
2.1. Regional Differences in Gene Expression
2.2. Top Differentially Expressed Genes (DEGs)
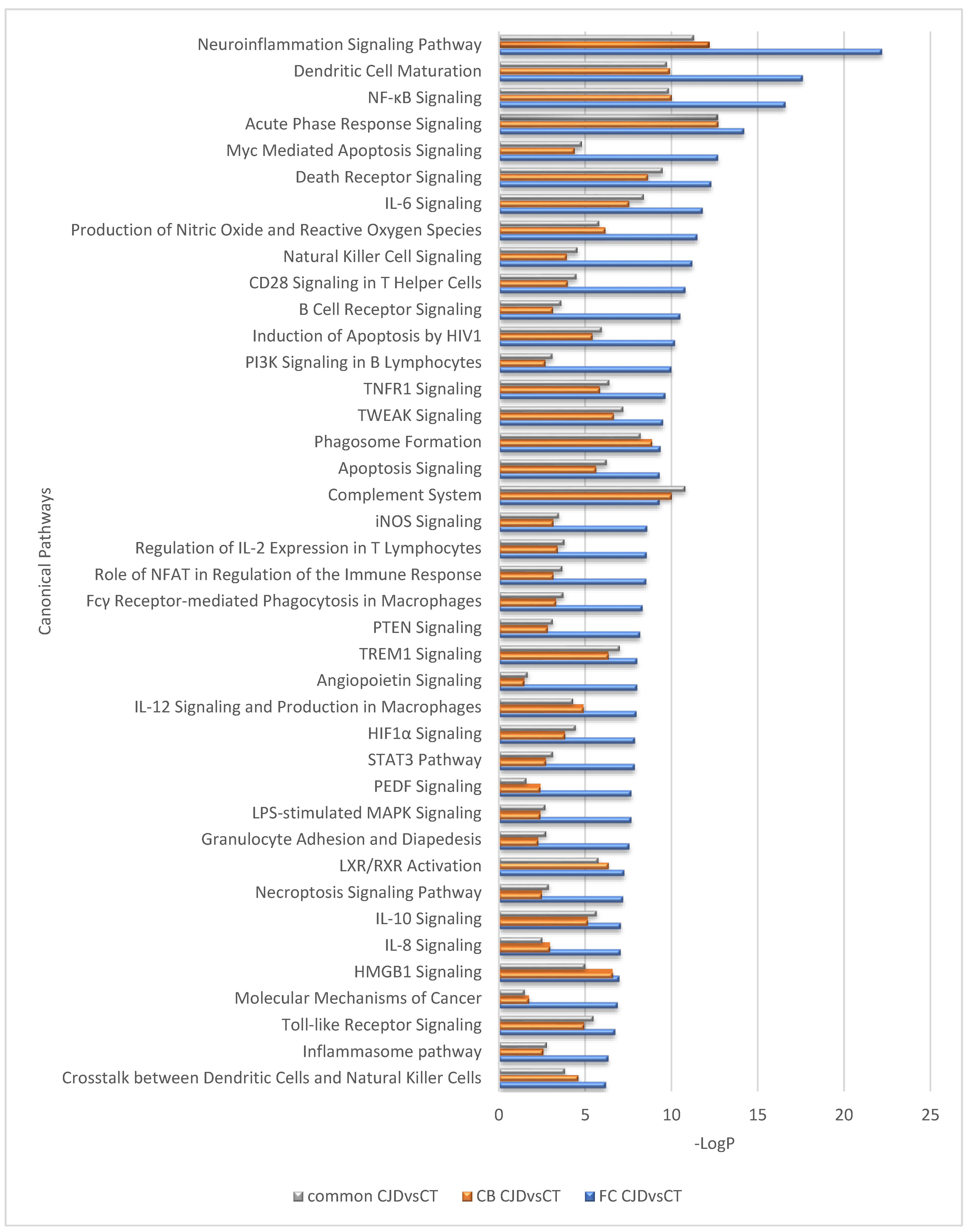
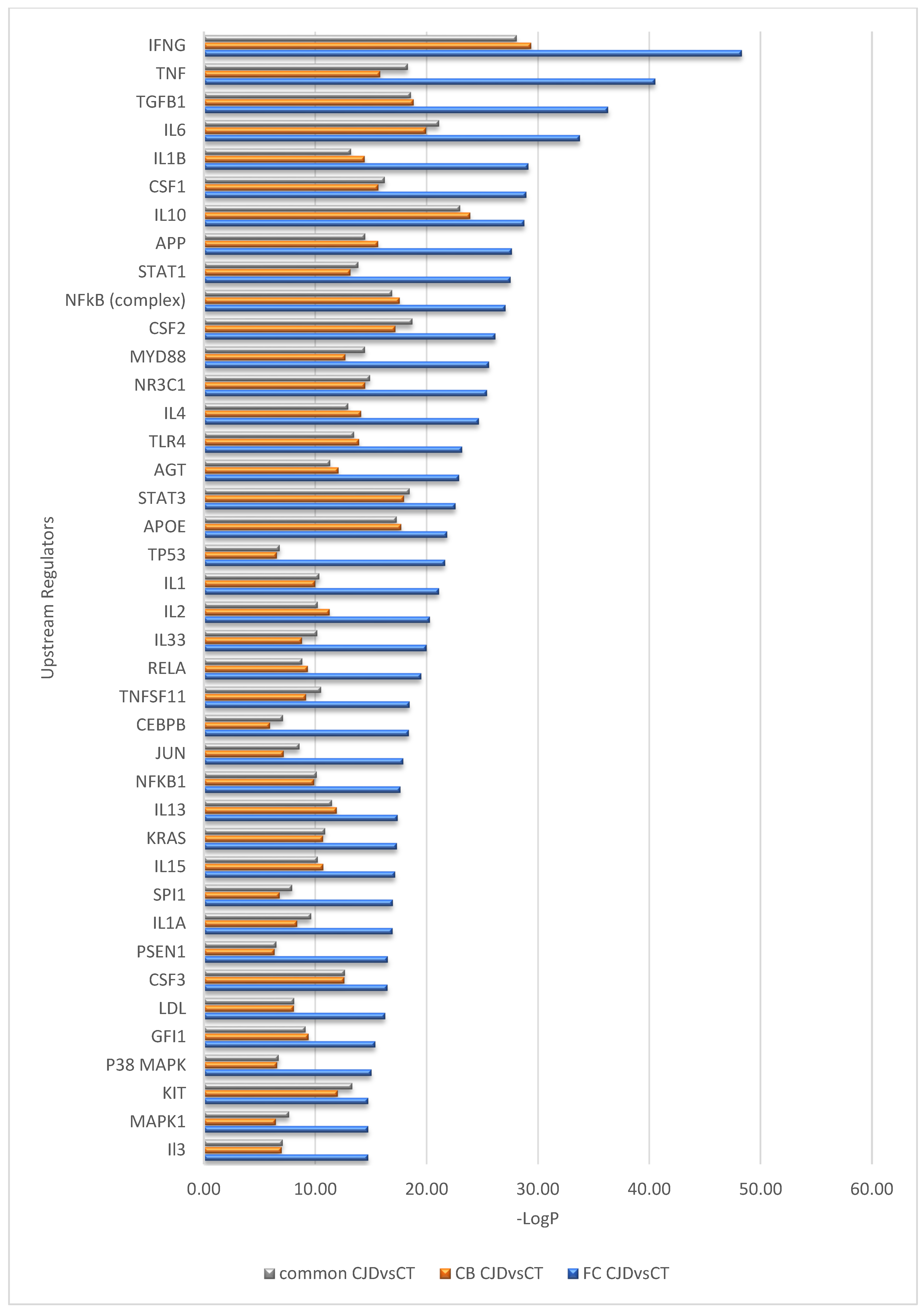
2.3. Inter-Regionally Overlapping Canonical Pathways and Upstream Regulators
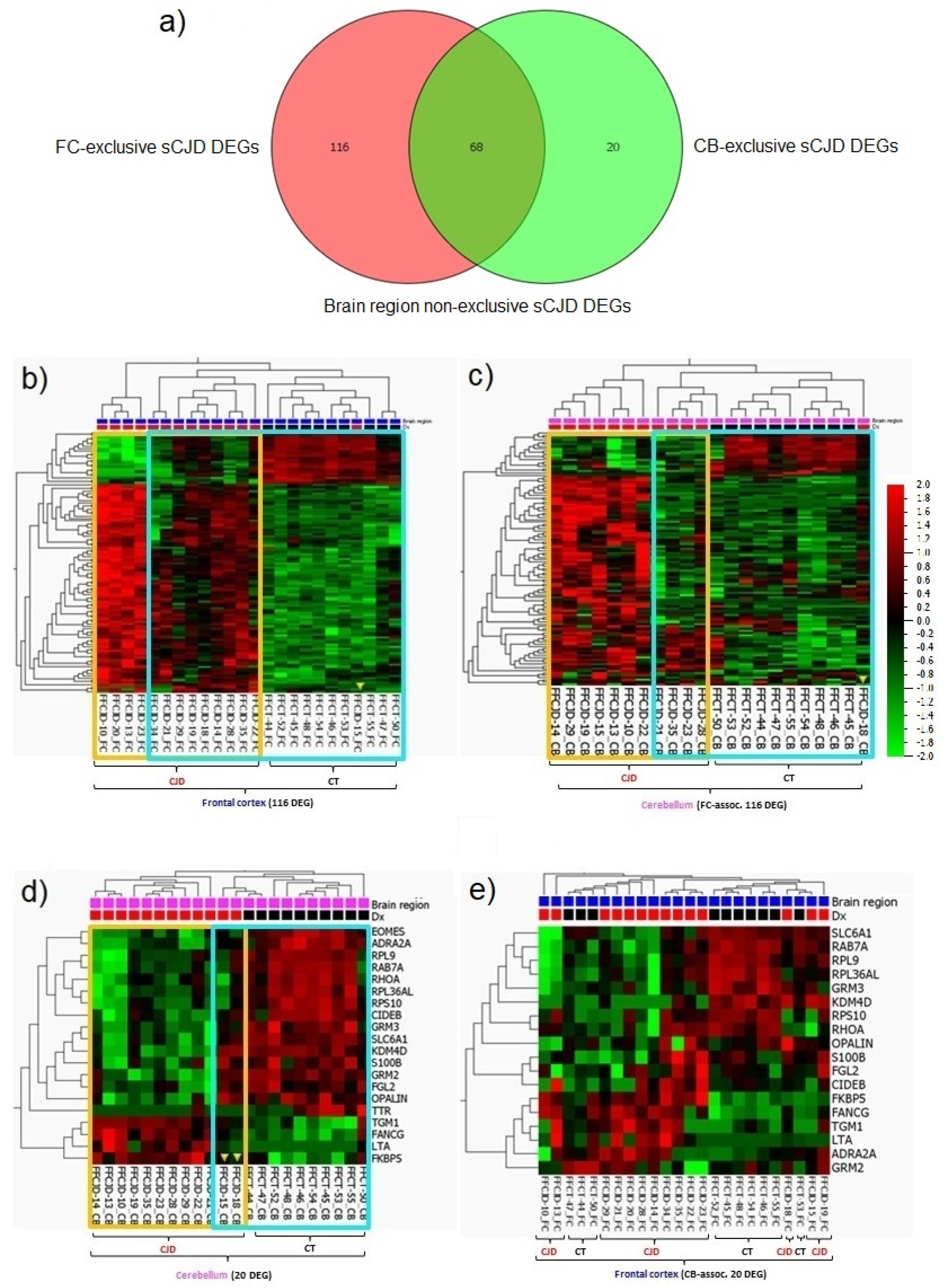
2.4. sCJD FC and CB-Exclusive DEGs
2.5. Sub-Regional Differences: Variance in the Strength of Neuroinflammation
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. RNA Extraction and Evaluation
4.3. NanoString
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| sCJD | Sporadic Creutzfeldt–Jakob disease |
| CT | Control tissue (normal brain) |
| FC | Frontal cortex |
| CB | Cerebellum |
| DEG(s) | Differentially expressed gene(s) |
References
- Prusiner, S.B. Biology and Genetics of Prions Causing Neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kitamoto, T.; Mizusawa, H. Iatrogenic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 207–218. [Google Scholar] [PubMed]
- Ladogana, A.; Kovacs, G.G. Genetic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 219–242. [Google Scholar] [PubMed]
- Zerr, I.; Parchi, P. Sporadic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 155–174. [Google Scholar] [PubMed]
- Areskeviciute, A.; Broholm, H.; Melchior, L.C.; Bartoletti-Stella, A.; Parchi, P.; Capellari, S.; Scheie, D.; Lund, E.L. Molecular Characterization of the Danish Prion Diseases Cohort With Special Emphasis on Rare and Unique Cases. J. Neuropathol. Exp. Neurol. 2019, 78, 980–992. [Google Scholar] [CrossRef]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Parchi, P.; Saverioni, D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol. 2012, 50, 20–45. [Google Scholar]
- Parchi, P.; Strammiello, R.; Giese, A.; Kretzschmar, H. Phenotypic variability of sporadic human prion disease and its molecular basis: Past, present, and future. Acta Neuropathol. 2010, 121, 91–112. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.M.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef]
- Franceschini, A.; Strammiello, R.; Capellari, S.; Giese, A.; Parchi, P. Regional pattern of microgliosis in sporadic Creutzfeldt-Jakob disease in relation to phenotypic variants and disease progression. Neuropathol. Appl. Neurobiol. 2018, 44, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.S. Selective vulnerability to neurodegenerative disease: The curious case of Prion Protein. Dis. Model. Mech. 2014, 7, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parchi, P.; De Boni, L.; Saverioni, D.; Cohen, M.L.; Ferrer, I.; Gambetti, P.; Gelpi, E.; Giaccone, G.; Hauw, J.-J.; Höftberger, R.; et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: An inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012, 124, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alibhai, J.; Blanco, R.A.; Barria, M.A.; Piccardo, P.; Caughey, B.; Perry, V.H.; Freeman, T.C.; Manson, J.C. Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration. PLoS Biol. 2016, 14, e1002579. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Crespo, I.; Roomp, K.; Jurkowski, W.; Kitano, H.; Del Sol, A. Gene regulatory network analysis supports inflammation as a key neurodegeneration process in prion disease. BMC Syst. Biol. 2012, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Investig. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.; Lee, I.Y.; Yoo, H.; Gehlenborg, N.; Cho, J.; Petritis, B.; Baxter, D.; Pitstick, R.; Young, R.; Spicer, D.; et al. A systems approach to prion disease. Mol. Syst. Biol. 2009, 5, 252. [Google Scholar] [CrossRef]
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2016, 90, 3003–3017. [Google Scholar] [CrossRef] [Green Version]
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.; Biber, K.; Michelucci, A. Cellular and Molecular Characterization of Microglia: A Unique Immune Cell Population. Front. Immunol. 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Ansoleaga, B.; Garcia-Esparcia, P.; Zafar, S.; Grau-Rivera, O.; López-González, I.; Blanco, R.; Carmona, M.; Yagüe, J.; Nos, C.; et al. PrP mRNA and protein expression in brain and PrPcin CSF in Creutzfeldt-Jakob disease MM1 and VV2. Prion 2013, 7, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens, F.; Lopez-Gonzalez, I.; Thune, K.; Carmona, M.; Zafar, S.; Andreoletti, O.; Zerr, I.; Ferrer, I. Subtype and regional-specific neuroinflammation in sporadic creutzfeldt-jakob disease. Front. Aging Neurosci. 2014, 6, 198. [Google Scholar] [CrossRef] [PubMed]
- López-González, I.; Garcia-Esparcia, P.; Llorens, F.; Ferrer, I. Genetic and Transcriptomic Profiles of Inflammation in Neurodegenerative Diseases: Alzheimer, Parkinson, Creutzfeldt-Jakob and Tauopathies. Int. J. Mol. Sci. 2016, 17, 206. [Google Scholar] [CrossRef] [PubMed]
- nCounter Neuroinflammation Panels. Available online: https://www.nanostring.com/products/gene-expression-panels/gene-expression-panels-overview/ncounter-neuroinflammation-panels (accessed on 20 December 2020).
- Bartoletti-Stella, A.; Corrado, P.; Mometto, N.; Baiardi, S.; Durrenberger, P.F.; Arzberger, T.; Reynolds, R.; Kretzschmar, H.; Capellari, S.; Parchi, P. Analysis of RNA Expression Profiles Identifies Dysregulated Vesicle Trafficking Pathways in Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2019, 56, 5009–5024. [Google Scholar] [CrossRef]
- Liu, S.; Hossinger, A.; Hofmann, J.P.; Denner, P.; Vorberg, I. Horizontal Transmission of Cytosolic Sup35 Prions by Extracellular Vesicles. mBio 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Liu, S.; Hossinger, A.; Göbbels, S.; Vorberg, I. Prions on the run: How extracellular vesicles serve as delivery vehicles for self-templating protein aggregates. Prion 2017, 11, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Makarava, N.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol. Dis. 2020, 137, 104783. [Google Scholar] [CrossRef] [PubMed]
- Fillman, S.G.; Cloonan, N.; Catts, V.; Miller, L.C.; Wong, J.; McCrossin, T.; Cairns, M.T.; Weickert, C.S. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2012, 18, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Chen, S.; Zheng, C.; Wei, H.; Song, X. Meta-Analysis of Gene Expression and Identification of Biological Regulatory Mechanisms in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin–protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Vanni, S.; Moda, F.; Zattoni, M.; Bistaffa, E.; De Cecco, E.; Rossi, M.; Giaccone, G.; Tagliavini, F.; Haïk, S.; Deslys, J.P.; et al. Differential overexpression of SERPINA3 in human prion diseases. Sci. Rep. 2017, 7, 15637. [Google Scholar] [CrossRef]
- Moody, L.R.; Herbst, A.J.; Yoo, H.S.; Vanderloo, J.P.; Aiken, J.M. Comparative prion disease gene expression profiling using the prion disease mimetic, cuprizone. Prion 2009, 3, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Striebel, J.F.; Race, B.; Phillips, K.; Chesebro, B. Prion Infection of Mouse Brain Reveals Multiple New Upregulated Genes Involved in Neuroinflammation or Signal Transduction. J. Virol. 2014, 89, 2388–2404. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Klaus, C.; Pricop-Jeckstadt, M.; Miller, J.A.; Struebing, F.L. A Microglial Signature Directing Human Aging and Neurodegeneration-Related Gene Networks. Front. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef]
- Baker, C.A.; Manuelidis, L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Booth, S.; Bowman, C.; Baumgartner, R.; Sorensen, G.; Robertson, C.; Coulthart, M.; Phillipson, C.; Somorjai, R.L. Identification of central nervous system genes involved in the host response to the scrapie agent during preclinical and clinical infection. J. Gen. Virol. 2004, 85, 3459–3471. [Google Scholar] [CrossRef]
- Bradford, B.M.; Wijaya, C.A.W.; Mabbott, N.A. Discrimination of Prion Strain Targeting in the Central Nervous System via Reactive Astrocyte Heterogeneity in CD44 Expression. Front. Cell. Neurosci. 2019, 13, 411. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, Y.; Li, H.; Li, Y.; Li, M.; Yuan, Y.; Cui, S.-S.; Yao, D. Effect of Spp1 on nerve degeneration and regeneration after rat sciatic nerve injury. BMC Neurosci. 2017, 18, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.-T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain, Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coscia, F.; Doll, S.; Bech, J.M.; Schweizer, L.; Mund, A.; Lengyel, E.; Lindebjerg, J.; Madsen, G.I.; Moreira, A.J.M.; Mann, M. A streamlined mass spectrometry-based proteomics workflow for large scale FFPE tissue analysis. J Pathol 2020, 251, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499. [Google Scholar] [CrossRef] [Green Version]
- Haigh, C.L.; Groveman, B.R.; Walters, R. Using our mini-brains: Cerebral organoids as an improved cellular model for human prion disease. Neural Regen. Res. 2020, 15, 1019–1020. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brain Region | Gene | Gene Expression | Molecular Function 4 | Biological Process 4 | |
|---|---|---|---|---|---|
| Group 1 DEGs 1 | Frontal Cortex & Cerebellum | SERPINA3 †, Serpin Family A Member 3 |  | The SERPINA3 protein inhibits serine proteases by binding to them, and thus inducing an irreversible conformational change; identical protein binding. | Acute-phase response; cellular protein metabolic process; endoplasmic reticulum to Golgi vesicle-mediated transport; neutrophil degranulation; post-translational protein modification; blood coagulation. |
| SOCS3, Suppressor of Cytokine Signaling 3 |  | Negatively regulates cytokine signal transduction; 1-phosphatidylinositol-3-kinase regulator activity; phosphotyrosine residue binding; protein kinase inhibitor activity. | Negative regulation of apoptotic process and inflammatory response, of receptor signaling pathway via JAK-STAT, and of tyrosine phosphorylation of STAT protein; positive regulation of cell differentiation; post-translational protein modification. | ||
| SPP1 †, Secreted Phosphoprotein 1 |  | Probably important to cell-matrix interaction; cytokine activity; extracellular matrix binding; integrin binding. | Cell adhesion; cellular protein metabolic process; inflammatory response; positive regulation of transcription, DNA-templated; post-translational protein modification. | ||
| CD44 †, CD44 Molecule |  | Cell-surface receptor that plays a role in cell-cell interactions, cell adhesion and migration, helping them to sense and respond to changes in the tissue microenvironment; collagen and hyaluronic acid binding. | Positive regulation of heterotypic cell-cell adhesion; cell migration; extracellular matrix disassembly; negative regulation of apoptotic process; inflammatory response. | ||
| FCER1G, Fc Fragment of IgE Receptor Ig |  | IgE-binding protein; receptor; identical protein binding; IgG binding. | Immunity; innate immunity; mast cell activation; phagocytosis, engulfment; positive regulation of interleukin-10, -6, and -4 production. | ||
| Group 2 DEGs 2 | Frontal Cortex | DAB2, DAB Adaptor Protein 2 |  | Adapter protein; cargo receptor activity; clathrin adaptor activity; protein C-terminus binding; SMAD binding. | Apoptotic process; cell differentiation; membrane organization; protein transport; negative regulation of protein binding and protein localization to plasma membrane; positive regulation of cell migration and early endosome to late endosome transport. |
| ITGB5, Integrin Subunit Beta 5 |  | A receptor for fibronectin; integrin binding; signaling receptor activity; virus receptor activity. | Cell adhesion mediated by integrin; cell migration; extracellular matrix organization; stress fiber assembly; host-virus interaction. | ||
| GRAP *, Growth Factor Receptor-Bound Protein 2-Related Adaptor Protein |  | Couples signals from receptor and cytoplasmic tyrosine kinases to the Ras signaling pathway. | Cell-cell signaling; Ras protein signal transduction; sensory perception of sound. | ||
| TGFBR1, Transforming Growth Factor Beta Receptor 1 |  | Serine/threonine-protein kinase; receptor; activin binding; ATP binding; metal ion binding; SMAD binding; transforming growth factor beta binding. | Apoptosis; differentiation; growth regulation. | ||
| TNFRSF10B *, Tumor Necrosis Factor Receptor Superfamily, Member 10b |  | Receptor for the cytotoxic ligand TNFSF10/TRAIL mediating apoptosis; promotes the activation of NF-kappa-B; essential for ER stress-induced apoptosis. | Apoptosis; cellular response to mechanical stimulus; leukocyte migration; response to endoplasmic reticulum stress. | ||
| Cerebellum | ADRA2A *, Adrenoceptor Alpha 2A |  | G-protein coupled receptor; transducer; protein heterodimerization activity; protein homodimerization activity. | Cellular response to hormone stimulus; glucose homeostasis; positive regulation of cell migration, cell population proliferation, cytokine production; Rho protein signal transduction. | |
| GRM2, Glutamate Metabotropic Receptor 2 |  | G-protein coupled receptor; transducer; may mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. | Chemical synaptic transmission; glutamate homeostasis and secretion; G protein-coupled glutamate receptor signaling pathway; regulation of synaptic transmission, glutamatergic. | ||
| RHOA, Ras Homolog Family Member A |  | Hydrolase; in neurons, involved in the inhibition of the initial spine growth. Upon activation by CaMKII, modulates dendritic spine structural plasticity. | Cell cycle; cell division; host-virus interaction; cerebral cortex cell migration; cellular response to chemokine and cytokine stimuli; forebrain radial glial cell differentiation; neuron apoptosis, morphogenesis, proliferation, and differentiation. | ||
| S100B, S100 Calcium Binding Protein B |  | Calcium-dependent protein binding; identical protein binding; protein homodimerization activity; tau protein binding. | Astrocyte differentiation; central nervous system development; innate immune response; regulation of cell shape; positive regulation of cell population proliferation and apoptotic process. | ||
| SLC6A1, Solute Carrier Family 6 Member 1 |  | Terminates the action of GABA by its high affinity sodium-dependent reuptake into presynaptic terminals; identical protein binding; metal ion binding; neurotransmitter binding. | Neurotransmitter transport; neurotransmitter reuptake; synapse organization; transport across blood-brain barrier. | ||
| Group 3 DEGs 3 | Frontal Cortex | ASB2, Ankyrin Repeat and SOCS Box Containing 2 |  | Mediates the ubiquitination and subsequent proteasomal degradation of target proteins. | Intracellular signal transduction; post-translational protein modification. |
| DLX1, Distal-Less Homeobox 1 |  | Transcriptional activator or repressor; plays a role in differentiation of interneurons, in the development of the ventral forebrain and diencephalic subdivisions, in craniofacial patterning and morphogenesis. | Cell differentiation; transcription; transcription regulation; developmental protein. | ||
| NRGN, Neurogranin |  | Acts as a messenger during synaptic development and remodeling; calmodulin binding; phosphatidic acid binding; phosphatidylinositol-3,4,5-trisphosphate binding. | Nervous system development; positive regulation of long-term synaptic potentiation; postsynaptic modulation of chemical synaptic transmission; signal transduction; telencephalon development. | ||
| Cerebellum | EOMES, Eomesodermin |  | Transcriptional activator; plays a role in brain development being required for the specification and the proliferation of the intermediate progenitor cells and their progeny in the cerebral cortex. | Adaptive immune response; brain development; cell fate specification; cerebral cortex neuron differentiation; cerebral cortex regionalization; stem cell population maintenance; interferon-gamma production. | |
| TTR, Transthyretin |  | Probably transports thyroxine from the bloodstream to the brain; hormone activity; identical protein binding; protein-containing complex binding; thyroid hormone binding. | Cellular protein metabolic process; extracellular matrix organization; neutrophil degranulation; purine nucleobase metabolic process. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Areškevičiūtė, A.; Litman, T.; Broholm, H.; Melchior, L.C.; Nielsen, P.R.; Green, A.; Eriksen, J.O.; Smith, C.; Lund, E.L. Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt–Jakob Disease Patients. Int. J. Mol. Sci. 2021, 22, 140. https://doi.org/10.3390/ijms22010140
Areškevičiūtė A, Litman T, Broholm H, Melchior LC, Nielsen PR, Green A, Eriksen JO, Smith C, Lund EL. Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt–Jakob Disease Patients. International Journal of Molecular Sciences. 2021; 22(1):140. https://doi.org/10.3390/ijms22010140
Chicago/Turabian StyleAreškevičiūtė, Aušrinė, Thomas Litman, Helle Broholm, Linea C. Melchior, Pia R. Nielsen, Alison Green, Jens O. Eriksen, Colin Smith, and Eva L. Lund. 2021. "Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt–Jakob Disease Patients" International Journal of Molecular Sciences 22, no. 1: 140. https://doi.org/10.3390/ijms22010140
APA StyleAreškevičiūtė, A., Litman, T., Broholm, H., Melchior, L. C., Nielsen, P. R., Green, A., Eriksen, J. O., Smith, C., & Lund, E. L. (2021). Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt–Jakob Disease Patients. International Journal of Molecular Sciences, 22(1), 140. https://doi.org/10.3390/ijms22010140