Chemical Bonding by the Chemical Orthogonal Space of Reactivity


Abstract
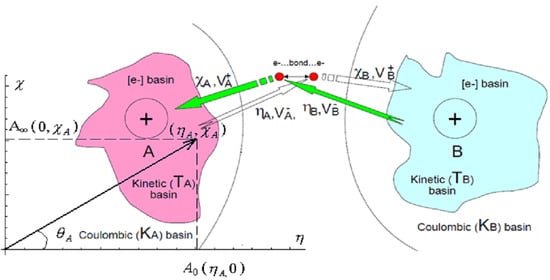
:
1. Introduction
- Electronegativity and chemical hardness actually furnish the most general principles of chemistry, due to their variational character [21,22], thus supporting both the equilibrium as well as the equilibrium fluctuation of isolated and interacting states, as reflected in the celebrated principles of equalization electronegativity, minimum electronegativity, maximum hardness, and hard and soft acids and bases [23,24,25,26,27];
- The atomic scales computed with electronegativity and chemical hardness generally parallel the other structural parameters’ dependencies, such as estimated by atomic radii [28], ionization potential [29], electronic affinity [30], diamagnetic susceptibility [31], polarizability [32], etc. Thus, they prove once more their structural and eventually observational features, despite the “many faces” of their appearance [33,34,35,36,37,38];
- The molecular use of electronegativity and chemical hardness are equally valuable: it spans from modeling the chemical bonding by HOMO-LUMO coupling through the application of their chemical reactivity principles (i.e., the above variational principles) [39,40,41], to the localization-delocalization of electronic characterization and bond sharing of atoms in molecules [42]. The purpose is to model another specific chemical concept as the aromaticity [43], to the use of their intriguing parabolic combination in the total/valence energy to provide the molecular reactivity hierarchies [44], to model the toxicity and the eco-, bio-, and pharmacological activities by the consecrated quantitative structure-activity relationships (QSARs), as well by the recent Quantum-SAR (Qua-SARs) [45,46]. It may also include the recent coloring framework of the topological approaches offered by chemical graph theory [47] either by discrimination on chemical bonding path in molecules, or by chemical bonding in adjacencies [48], this way succeeding in discriminating for the chemical conjugation and providing the unified treatment of kenograms and pleograms [49], while offering new development space in chemical graph theory [50];
- Electronegativity and chemical hardness are also used as the benchmark qualitative framework in providing the oxidation and reduction potentials in electrochemistry models and in the design of efficient electro-photovoltaic cells, with great insight in selecting the appropriate sensitizer on anodic electrode, especially in the current modeling of the third generation of photosensitizes based on quantum-dots technology [51,52].
- Section 2 introduces the chemical power concept in the chemical orthogonal space this way conceptually and analytically linking the electronegativity and chemical hardness by their ratio, considering it as the precursor of the charge transfer atoms-in-bonding expression. The homo- and heteronuclear atoms-in-bonding cases follow naturally as a generalization of PP former expression, while the conceptual–analytical advantage of the present approach is revealed in the homonuclear bonding case by surpassing the limitative zero charge transfer and energy in such bonding and elegantly avoiding the un-physicochemical picture of recording the redundancy of electronegativity and chemical hardness at both atomic and molecular levels;
- Section 3 deals with the application of the present atoms-in-bonding formalism for the halogen acids series of hetero- and homonuclear diatomic systems and the produced results are analyzed with the available experimental data both numerically and graphically. Finally, the tricky problem of molecular fragmentation and its reverse problem like the establishing of molecular pattern formation by its fragments and atomic groups is analyzed using the successive ad-atom methodology. To it we added the maximum chemical hardness jointly with minimum chemical power hierarchy to establish the most probable molecular channel formation by recursive atoms-in-bondings in polyatomic molecules, which were chosen as the prototype chemical ones (H2O, NH3, CH4, and C6H6).
2. Conceptual Method
2.1. Orthogonal Space of Chemical Reactivity: The Chemical Power Concept
- electronegativity driving the tendency of the system in achieving electrons (so manifesting the reverse “chemical potential”); and
- chemical hardness controlling the tendency to inhibit the bonding and atoms-in-molecule coordination, being related with the system’ s chemical stability (so manifesting the “chemical inertia/force”).
2.2. The Case of Heteronuclear Bonding (in Diatomic Molecules)
2.3. The Case of Homonuclear Bonding (in Diatomic Molecules)
2.4. Parr–Pearson vs. COS Approaches
- There is a general rule that larger molecules are chemically less harder (or softer) along congeners, when electronegativity is reduced or enhanced depending the substituent type; for example χ is decreasing by metal and mercapt on one side, and increased by phenyl, respectively, with direct consequence on physicochemical properties as the solvation free energy, or absorptivity on a surface, corrosion, etc., for nanomaterials [60];
- The reaction mechanism of functional groups, especially in organic chemistry, may be considerably understood when considering the electronegativity relationship with charge transfer and energy of atoms-in-molecules; see, for instance, the group electronegativity as well as the derived equations from the Sanderson equalization principles eventually applied to ionic groups [61];
- Chemical hardness provides a fruitful route for developing the density functional softness theory with the allied hierarchy for kernel, local, and global (including spin) reactivity indices, eventually leaving with new formulation of the linear response functions so useful in modeling the reactivity patterns of (open shell) molecules [62];
- The fashioned expressions of electronegativity and chemical hardness in their spectroscopic forms, i.e., 0.5 (I + A) and 0.5 (I − A), respectively, relay on the second order cutoff of the atomic energy Taylor expansion when ionization energy obeys the dependency In = nI1, with a wide applications on the A-Groups II, IV–VIII, excepting the oxygen [63];
- The electronegativity as the complementary chemical hardness influence determines also the super-molecule model of solute–solvent interaction and charge transfer, within the continuum polarizable charge model; actually, as the electrostatic potential diminishes by increasing of the effective radii of neutral and charged solute, the electronegativity equalizes between the solute and solvent and the chemical hardness diminishes too, so enhancing the chemical reactivity induced by solvent effects [64];
- Even the unconventional exohedral fullerenes as C64X4 (X = H, F, Cl, Br, and I) are subjected to chemical reactivity analysis through the basic electronegativity and chemical hardness indices: they both decrease with increasing atomic number of X in C64X4 cluster molecules. Instead, the CX fragment has electronegativity dependent on its located site. However, the electrophilicity index (ω = χ2/2η) increases from C64F4 to C64I4 paralleling the decreasing stability recorded by means of decreasing of bond dissociation energies, energy gaps, and maximal frequencies. They indicate a general nonaromatic character for the carbon halogen molecules of C64X4 (X = F, Cl, Br and I) [65];
- Electronic properties of oxides were successfully established in terms of the electronegativity- chemical hardness binomial (χ, η) coupling namely as (i) (χ↑ & η↓) is specific for oxides of transition elements with high oxidation state; (ii) (χ↑ & η↑) characterizes the insulators with large optical (HOMO-LUMO) gap; (iii) (χ↓ & η~>) feature alkali and alkaline earth oxides; and (iv) (χ↓ & η↑) describe ionic oxides with relatively large optical gap [66];
- The atoms-in-molecule Bent rule, according to which “more electronegative substituents prefer hybrid orbitals having less ‘s’ character and more electropositive substituents prefer hybrid orbitals having more ‘s’ character” was found consistent with the maximum chemical hardness reactivity principle, for a series of isomers, since the more electronegative atom occupies the axial position has maximum hardness. However, the same rule is less correlating with the minimum polarizability principle, being the last more sensitive to the basis set used, especially when the diffusion is this way better represented; application on different isomers of SF4, SF4O as well as for a family of PClxF5−x (x = 1–4) in combination with B3LYP with different basis sets of computational density functional theory confirms such general tendencies [67];
- On the other side, the simple MP2 model combined with the straight approximation of chemical hardness kernel as the Dirac localized function, , finely orders the Lewis acids and bases the computed global chemical hardness perspective when compared with experimental data [68].
- The AB bonding case of COS consistently recovers the molecular AA case of Parr–Pearson;
- At the AA bonding level, the COS and PP approaches become identical when the chemical hardness and electronegativity interchanges their role by Equation (32b); this is in agreement with the reduction of the Figure 4 to Figure 3, so consecrating the present orthogonal chemical reactivity approach with the internal consistency (see also the forthcoming numerical applications in Section 3);
3. Results and Discussions
- The standard B3LYP/6-311G++(d,p) method along the basis set calculation within density functional theory for reactivity indices as ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical hardness (η), and the electrophilicity index (ω) have proved to correlate with HOMO and LUMO energies. They relate the molecular spectroscopic properties such as vibrational spectral analysis, e.g., carried out by Raman and infrared spectroscopy in the range 4000–400 cm−1 and 3500–100 cm−1, respectively, for the 2-hydroxy-5-bromobenzaldehyde (HBB), a benzaldehide derivative with potential action in anti-microbial activity ant-cancer properties, to reveal that the charge transfer mainly occurs within the molecule, as based on the produced electrostatic potential maps [70];
- Natural bond orbital analysis (NBO, NBA) may be carried out for establishing the charge transfer between the localized bonds and lone pairs; their calculated electronic and optical properties, i.e., absorption wavelengths, excitation energy, dipole moment, molecular electrostatic potential (MEP), and frontier molecular orbital energies correlate with the observed X-ray diffraction, FT-IR, and UV-Vis spectroscopically methods; as a result, the elucidation of the intramolecular hydrogen bonded interactions may be established, for instance, strong O–H···N interactions in enol-imine form and N–H···O interactions in keto-amine for Schiff base compounds (such as (E)-2-((4-hydroxy-2-methylphenylimino)methyl)-3-methoxyphenol) exhibit unique properties in catalysis and medicinal chemistry [71];
- Schiff bases generally display biological activity superior to the free ligands, with direct application to the study of DNA binding, cytotoxicity and apoptosis. Therefore, they become viable alternative for common chemosensors used in molecular recognition and anion sensing; this is due to the manifested tautomerism of the OH group in ortho position as well the imino group in both solution and solid states, especially at the level of the hydrogen bonds O-H···N or O···H-N between the enol-imine and keto-amine forms, respectively [72];
- Charge reversal of cations (+CR−) stands as a complementary method in the arsenal of mass spectroscopy methods used in distinguishing the prototype hydrocarbon cations, such as C6H5+ and C6H6+●; the last compound is probably the most studied ion by coincidence measurements, collision- and surface-induced dissociation, charge-stripping, neutralization–reionization, and ion–molecule reactions spectrometric methods [73];
- The charge transfer may be considered to be the driving parameter for the adatom–metal bond strength, thus anticipating the adsorption energy trends in terms of electronegativity and chemical hardness. The work function is used for the metal surface electronegativity, with the electronegativity of metal being a combination of the atomic Mulliken electronegativity and of the work function of the metal surface. The resulting charge transfer is viewed as the metal surface and adatom electronegativity difference These results further consecrate the HSAB principle in terms of charge transfer parameter ΔN [74].
3.1. Diatomic Molecules with Hetero- and Homo-Atoms
3.2. Atomic Paths in Molecular Formation
- The resolution mass spectroscopy investigates the mechanisms through which fragment ions are formed; in particular, the remote site fragmentation with charge localization can be useful for identifying ions containing portions of molecule, e.g., ammonia chemical ionization for cardiac glycosides [77];
- Fourier transform mass spectrometry may indicate the rapid adduct formation through extrusion reactions, e.g., the direct extrusion of CO from furan, or HCN extrusion from pyrrole, by the aid of hydrogen-atom displacement dominant reaction, eventually followed by additional neutral loses [78];
- Molecular fragmentation may be produced by ionic-molecular collision at keV projectile energies, while the range of eV corresponds to the Coulomb explosion of ionized target, e.g., (H2O)q+ in He2+ +H2O reaction, sometimes with smaller absolute cross section than the classical Rutherford formula, due to the screening of the bound electrons [79];
- Atoms-in-molecule properties, such as zero-point kinetic energy, may be determined by high-resolution electron Compton scattering combined with the Doppler broadening recording of the atomic momentum distribution in molecule contributing to both external and internal molecular motion, respectively; e.g., the e-scattering intensities from H- and O- atoms in molecular systems such as H2, CH4, H2O, and NH3 [80];
- Molecular fragments may also have an important role in selective catalytic reduction, eventually monitorized by the spectroscopic techniques such as an in situ FT-IR spectrophotometry; e.g., the case of NHx species production by the introduction of H2 into the reduction of NO over Pt-MnOx catalysts [81];
- The pressure effect on the molecular fragment dynamics over the catalyst is especially present in the final thermodynamically part of the reaction, approaching its equilibrium, and not in its first part when the kinetics is dominant by the mass transfer and consumption of the O2 and fuel; e.g., the case of catalytic partial oxidation of light hydrocarbons, e.g., the CH4 and C3H8, eventually leading with the intermediate species C2+ [82];
- Bifunctional molecules (e.g., CH3OCH2COCH3 and CH3COCOCH3) are in general found to be less stable in low temperature solids as compared with the simple generic compounds, by the so called “hot” fragmentation with a probability decreasing with the excess energy diminishment, down group of periodic system. This is the case of solid rare gas matrices, e.g., yielding radicals upon the irradiation in solid Ar matrix at T < 16K [83];
- Preferential breaking of the strain bonds by laser ablation is an effective experimental method for controlling the molecular fragmentation, especially on good surface heating in the absence of any direct absorbance on the surface by from the concerned strained compound, e.g., ring compounds deposited on metal rods [84];
- Ligand elimination and ligand decomposition channels can also be approached by laser vaporization through laser photo-dissociation; even in exotic complexes such as bare uranium cations U+(C6H6)n=1–3 and uranium oxide ions UOm+(C6H6)m=1,2, the first one is more effective in expelling the neutral benzene in a ground state from the excited U+, U+(C2H2), and U+(C4H2) fragments’ decay, once they are formed under ultraviolet action on the former uranium complex [85];
- The main molecular dissociation channels may be established both by density functional calculation as well as from experimental electrospray mass spectroscopy fragmentation spectra; the consecrated example is that of protonated uracil (UH+) fragmentation using collision at increased energy, leading mainly to extraction of isocyanic acid (HNCO, 43Th) from the aromatic (cationic uracil) cycle, and yielding the C3H3NO fragment of 69-Th by means of the retro Diels-Alder reaction mechanism [86];
- Approaching the identification of the fragmentation mass spectra that are not contained in spectral library is under current focus in order to overcome the limits of the “known universe in organic chemistry” with huge consequence in assessing the genomics, proteomics, and metabolomics open issues. To this aim the extensive computational approach is replaced with similarity and fitting studies while having the molecular structure and allied reactivity properties such as reactivity indices of electronegativity and chemical hardness in their forefront [87].
- Conceptual-computational methods are truly effective in establishing the molecular inverse problem, i.e., determining the parent molecule (when searching for elucidation of structure of new/unknown materials) by simulated fragmentation pathways; for instance, the Simplified Molecular Input Line Entry Specification (SMILES) correlates satisfactorily with peak evolution in Gentle Secondary Ion Mass Spectrometry with fragmentation pathway mapping (G-SIMS-FPM) methods, as was essentially found for simulated pathways for amino acids and simple peptide [88]. A similar method was used also for studying the folic acid (with its six subunits: α, β, γ, δ, ε, ζ) and Irganox 1010 (a central carbon atom surrounded by four equal side-chains) through varying the G-SIM surface plasma temperature aiming producing new data-based system (including amino acids and simple peptides). The acquisition of fragmentation pathway simulation/molecular structure re-assemble information (about 90% of the fragments explained) plays a major role in analyzing the interaction in bio-molecule (protein) surface by means of polymer, drug-delivery, and organic electronics technologies [89];
- The synergy of atoms-contributing-catalysts as are Ni and Cu in NiO formation by plasma-treated sample of Ni-CU/Al2O3 nanocatalyst features enhanced reactivity compared with just impregnated nanocatalysts, since more uniform morphology recorded (e.g., by XRD and TEM techniques) for the first case, not withstanding that these nanomaterials lose about 10–12% of CH4 and CO2 through conversion, respectively, during the overall time of stream test due to the methane decomposition and decrease the RWGS (reverse water gas shift) reaction rate, while gas hourly space velocity (GHSV = Reactant Gas Flow Rate/Reactor Volume) has less effect on the reforming reactions for plasma-treated sample due to well-defined morphology of the nanocatalyst [90];
- Subtle aspects of energetics of intra- and inter- fragment density rearrangements, charge transfer and orbital mixing was recently observed through computational experiments of Wernerian complexes in the bonding regime of ligand field effects allowing new quantum insight in understanding the ligand field stabilization energy (LFSE); the method involves the energetic decomposition of the metal ion by ligand sets with fractional charges as resulted upon preliminary electronegativity equalization effect driven by charge transfer, thus producing their separate nominal oxidation states. This way, the 10Dq separation for prototype octahedral units (such as [MqF6]q−6, [Mq(CN)6]q−6, and [Mq(H2O)6]q complexes with MII and MIII ions selected from the M = Cr to Cu 3d series) was predicted by LFSE and electron promotion effects, leading to the interesting interplay between the ionic and covalent bonding regimes in coordination bonding characterization [91];
- The metal-benzene bonding clusters, i.e., Fe4–(C6H6)m, m ≤ 3, were characterized through metal (iron)–carbon bonding, driving the contour plots of molecular orbitals, while noting some isolated forbidden IR modes nearby those of isolated benzene; yet, IR activated ligand regime, as due to the 2p-π electrons of benzene interacting with the 3d electrons of Fe4 estimates ionization energies, electron affinities (so combined into electronegativity) and (related) binding energies in good agreement with experimental data [92];
- Fragmentation of carbon based molecules (such as glycine C2H5NO2) by slow (low-energy) monoenergetic electrons have been investigated with particular focus on the mechanism of formation of the doubly charged fragment (e.g., CH2NHCO2+); unusually, they are not detected by mass spectroscopy, since geometrical rearrangement of the initial molecule; however, they are accompanied by the C-H bond breaking with yield of the [OH + H] fragment [93]; the same mechanism and doubly charged fragment ionic formation was advanced also for methionine (C5H11NO2S) molecule with the only difference that the main channel of dissociation involves the sulfur atom, eventually leaving with the formation of the CH4S+, while the dominant channel formation of the C2H5S+ ionized fragment favors the charge stabilization of the sulfur atom [94];
- The mass analyzed threshold ionization (MATI) technique is another spectroscopic tool used for assessing the molecular fragmentation structure, especially for molecules with biological relevance by their manifested π-hydrogen bonded clusters, e.g., localization of the amino hydrogen in the pyrole ring of 3-methylindole·C6H6 cluster (58 018 cm−1) is comparable with the results obtained for 3MI·C6H6 cluster, thus confirming the methylation influence on the π-hydrogen bonding [95];
- The ultraviolet dispersed fluorescence spectroscopy was employed to provide the photo-fragmentation of H2O for photon excitations over 20eV; the identification of channel transition A2Σ+→X2ΠΩ for the OH fragment at higher energy about 30eV is in good agreement with vertical Rydberg states of water, but overcoming the dissociation limit for the dissociation channel OH(A2Σ+) + H*(n ≥ 2) [96].
- (i).
- Given a poly-atomic molecule, or a complex chemical system, the various add-atomic and add-bonding recursive combinations are considered in various channels patterning the final target molecule;
- (ii).
- Since the stabilization of the molecule is described by the maximum chemical hardness principle, the chemical hardness hierarchy is considered for the various channels in stage (i). It is nevertheless estimated through recursive pairs of add-atoms and add-bonds in the pursued patterns;
- (iii).
- The maximum chemical hardness principle is further combined with chemical power information, for which the associated minimum variational principle shall be applied: as based on the Equation (13), the chemical power contains also the electronegativity information, and of its minimum variation around equilibrium too, for the same channels patterning as analyzed in previous steps (i) & (ii).
3.3. Open Issues
- Their interrelation through the basic definitions as variation respecting the charge exchange in a reactive/boning system;
3.3.1. Computational Context for Generating Parameters
3.3.2. Relation with Aromatic Systems
3.3.3. Relation with Modern Valence Bond/Natural Resonance Theory
3.3.4. Connection with the van der Waals Potentials
3.3.5. Relation with the Bader’s Atoms-in Molecule’s Critical Bond
4. Conclusions and Perspectives
- Matter-intense X-ray interaction may spectroscopically resolve the bio-molecular fragmentation and production of the high charge atomic ions aiming the bio-imaging techniques at the femtosecond X-ray regime (pump and probe). It eventually may use a synchrotron free electron laser source to investigate multiple core-ionization-Auger decays by photoionic spectroscopy [109];
- The attosecond pulses spectroscopy techniques includes the generation of X-UV light sources, X-UV + IR pulses, eventually in combination with probe-pulse and steering of moving electrons, along with photoionization time delay. They reveal the electronic dynamics in a few electrons (i.e., ionized), atoms, and molecules in various fundamental (e.g., studying the quantum wave–particle duality) and the experimental (observing or predicting fragmentation) circumstances [110];
- Visualization of photons and induced many-particles fragmentation, similar with the bubble chamber in nuclear physics, may be achieved in the eV and mili-eV regimes through the reaction microscope (imaging) techniques, e.g., Cold Target Recoil Ion Momentum Spectroscopy = COLTRIMS, along the scanning tunneling microscopes, as part of many-particle sub-atomic physics and molecular fragment dynamics [111];
- HOMO and LUMO directly relate with charge transfer occurring within the molecule, a fact established also by Fourier Transform (FT)-IR and Raman spectra. The hyperconjugation and charge delocalization models are in agreement with natural bond orbital analysis (NBO); equally, the correlations of the variational spectra with the calculated potential energy distribution (PED) and with the chemical reactivity indices (among which electronegativity and chemical hardness are preeminent measure of stability and reactivity) provide reliable results [112];
- The question of whether the electronegativity information, once inserted in the dipole derivatives and the hardness’ Hessian of molecular energy, may provide sufficient or relevant correlation or prediction with/of spectroscopic data, e.g., within IR spectroscopy, eventually through polarizable and reactive force fields, was responded in negative [113]. Therefore, electronegativity requires supplementary information as is the present chemical orthogonal space (COS), where coupling with the chemical hardness is synergistically (complementarily yet simultaneously) developed;
- Elucidation of probe mechanism of action, including reactions of metal ions and organic compounds in aqueous solutions, onto solid surfaces, and for biological activity, may be unitarily treated with the so called four-element approach of quantum chemical reactivity theory: it customarily involves (i) electronic flow driven by electronegativity, (ii) polarization by condensed local softness (i.e., the inverse of chemical hardness), (iii) electrostatic interaction by atomic partial charge exchange, and (iv) hydrophilic interaction by the inverse of apolar surface area (1/APSA) [114];
- The relationship between the Pauling difference electronegativity and energy of heteronuclear dissociations was rationalized by means of two related concepts of the electronegativity and chemical hardness/softness through a two-variable (x,y) in an absolute space of variation, thus anticipating the present COS analysis and allowing the unitary homo- and heteronuclear treatment. While “x” directly relates with Pauling electronegativity, “y” quantifies the atomic intrinsic potential, thus relating with the atomic size contribution and depending on the contributing valence orbitals, etc. [115];
- Electronegativity may correlate with the hardness of crystalline materials, such as sphalerite, wurtzite, rocksalts, oxides α-SiO2 and LaGaO3, and graphite, as well for B12 analogs, group IVA nitrides, tungsten carbide materials, and transition metal di- and tetra-borides. In general, the rule that similar crystal structures associate with similar hardness anisotropy is developing, while establishing that the greater bond ionicity correlates with more orderly bond arrangements in single crystals [116];
- Fragmentation of nanostructures, from polycyclic aromatic hydrocarbon molecules (PAHs) to fullerenes, highly depends on collision energies, usually ranging from few tens to few hundreds of eV, while expelling single atoms; the dominance of bonds’ fragments of C2- or as C2H2-molecules (or H-atoms) released from fullerenes and PAHs is recorded, respectively. However, the C60 clusters may feature enhanced reactivity over the van der Walls range of fullerene molecules when releasing single C-atoms by producing C59+ reactive fragments, at their turn bonding covalently with another C60 molecule from the remaining cluster [117];
- Detection and identification of the large ion fragmentation used in bio-imaging (e.g., for peptides) may use the gas phase coupling surface-induced dissociation (SID) in a Fourier transform ion cyclotron resonance mass spectrometer with resonant ejection of selected fragment ions using a relatively short (5 ms) ejection pulse. It is supported by the Rice–Ramsperger–Kassel–Marcus (RRKM) theory prescribing that the shape of kinetic plots follows the shape and position of the energy deposition function specific to the internal energy distribution (and thus also to the entropy) for the ion-surface collision, the deciding to (identify the) most probable reaction channels [118];
- The solid oxide fuel anode may be sensitized to produce electricity (for more than 72h), H2O and CO2 by electrochemical oxidation of CH4 catalyzed by the perovskite lanthanum strontium cobalt ferrite through the basic processes as (i) decomposition of CH4, (ii) electrochemical oxidation of H to H2O, and (iii) electrochemical oxidation of C to CO2 with the formation rate greater than that for CO [119].
Funding
Informed Consent Statement
Conflicts of Interest
Appendix A. Quantum-Like Physical Formalization and Chemical Implications of COS
Appendix A.1. Add-in-Bonding Chemical Scattering Paradigm
- electronegativity (χ), as a measure of attraction of electrons to a system (and associating therefore with “creation action: ”, of a particle/electron by such effect), and to
- chemical hardness (η), measuring the inertia/resistance of a chemical system to the reactivity (so associating with the “annihilation action: ” process for a charge transfer effect).

- The system A may attract an electron from bonding by potential; at the same time, the system A may resist (“refuse”) the second electron from bonding since it repels with the first electron in bonding, through the repulsive action potential ;
- The same is valid also for the system B, developing the potentials for attracting one electron from bonding and repelling to the other, respectively.
- it normalizes the mixed potentials of chemical reactivity, so it appears in the denominator;
- it is written as four times the resistance of the mixed contribution to the bonding-by-reactivity potential (see above the Pauling resonance argument);
- such mixed resistance to bonding/reactivity corresponds to the chemical hardness kernel.
Appendix A.2. Hard-and-Soft-Acids-and-Bases (HSAB) Principle’s Verification
- a soft base, e.g., R− or H−, is very polarizable and thus with low electronegativity;
- a hard base, e.g., OH−, is not much polarizable and thus with high electronegativity (respecting the preceding soft base case);
- a soft acid, e.g., RO+ or HO+, has usually low positive charge and large size, so posing lower electronegativity;
- a hard acid, e.g., H+ or XH (hydrogen bonding molecules), has normally high positive charge and small size, so featuring high electronegativity (respecting the preceding soft acid case).

- The (s-s) case corresponds to the covalent bonding case and requires higher charge transfer ();
- The (h-h) case, paradoxically, does not furnishes high charge transfer (), yet being this way consistent with ionicity character of bonding by employing the same operational definition for ;
- The (s-h) and (h-s) cases should not favor chemical reactivity by lowering charge transferred quantities ().
- For (s-s) case: meaning that COS is more appropriate for covalency characterization;
- For (h-h) case: resulting COS as better description for ionicity character;
- For (s-h) and (h-s) cases: so affirming COS as less allowing the chemical reactivity by charge transfer, as the general prescription indicates.
References
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Pearson, R.G. Chemical Hardness; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Putz, M.V. Quantum Nanochemistry. A Fully Integrated Approach (5 Volumes Package): Vol. 1. Quantum Theory and Observability; Vol. 2. Quantum Atoms and Periodicity; Vol. 3. Quantum Molecules and Reactivity; Vol. 4. Quantum Solids and Orderability; Vol. 5. Quantum Structure-Activity Relationship (Qu-SAR); Apple Academic Press: Toronto, ON, Canada; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3808. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Putz, M.V. Quantum and electrodynamic versatility of electronegativity and chemical hardness. In Quantum Frontiers of Atoms and Molecules; Putz, M.V., Ed.; NOVA Science Publishers: New York, NY, USA, 2011; pp. 251–270. [Google Scholar]
- Putz, M.V. Density functionals of chemical bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Lee, H.; Parr, R.G. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar]
- Chattaraj, P.K.; Liu, G.H.; Parr, R.G. The maximum hardness principle in the Gyftpoulos-Hatsopoulos three-level model for an atomic or molecular species and its positive and negative ions. Chem. Phys. Lett. 1995, 237, 171–176. [Google Scholar] [CrossRef]
- Parr, R.G.; Bartolotti, L.J. On the geometric mean principle of electronegativity equalization. J. Am. Chem. Soc. 1982, 104, 3801–3803. [Google Scholar] [CrossRef]
- Parr, R.G.; Bartolotti, L.J. Some remarks on the density functional theory of few-electron systems. J. Phys. Chem. 1983, 87, 2810–2815. [Google Scholar] [CrossRef]
- Bergmann, D.; Hinze, J. Electronegativity and charge distribution. Struct. Bond. 1987, 66, 145–190. [Google Scholar]
- Mulliken, R.S. A new electroaffinity scale: Together with data on valence states and an ionization potential and electron affinities. J. Chem. Phys. 1934, 2, 782–793. [Google Scholar] [CrossRef]
- Putz, M.V. Electronegativity: Quantum observable. Int. J. Qual. Chem. 2009, 109, 733–738. [Google Scholar] [CrossRef]
- Putz, M.V. Chemical Hardness: Quantum Observable? Studia Universitatis Babeş-Bolyai—Seria Chemia–Tom I 2010, 55, 47–50. [Google Scholar]
- Koopmans, T. Uber die Zuordnung von Wellen Funktionen und Eigenwerter zu den Einzelnen Elektronen Eines Atom. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Putz, M.V. Koopmans’ analysis of chemical hardness with spectral like resolution. Sci. World J. 2013, 348415. [Google Scholar] [CrossRef] [Green Version]
- Komorowski, L. Electronegativity and hardness in chemical approximation. Chem. Phys. 1987, 55, 114–130. [Google Scholar] [CrossRef]
- Putz, M.V. Systematic formulation for electronegativity and hardness and their atomic scales within density functional softness theory. Int. J. Quantum Chem. 2006, 106, 361–389. [Google Scholar] [CrossRef]
- Ayers, P.W.; Parr, R.G. Variational principles for describing chemical reactions: The Fukui function and chemical hardness revisited. J. Am. Chem. Soc. 2000, 122, 2010–2018. [Google Scholar] [CrossRef]
- Ayers, P.W.; Parr, R.G. Variational principles for describing chemical reactions: Reactivity indices based on the external potential. J. Am. Chem. Soc. 2001, 123, 2007–2017. [Google Scholar] [CrossRef]
- Sanderson, R.T. Principles of electronegativity Part I. General nature. J. Chem. Educ. 1988, 65, 112–119. [Google Scholar] [CrossRef]
- Tachibana, A.; Nakamura, K.; Sakata, K.; Morisaki, T. Application of the regional density functional theory: The chemical potential inequality in the HeH+ system. Int. J. Quantum Chem. 1999, 74, 669–679. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases—The evolution of a chemical concept. Coord. Chem. Rev. 1990, 100, 403–425. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electronic structure principles and aromaticity. J. Chem. Edu. 2007, 84, 354–358. [Google Scholar] [CrossRef]
- Putz, M.V. Chemical action concept and principle. MATCH Commun. Math. Comput. Chem. 2011, 66, 35–63. [Google Scholar]
- Ghanty, T.K.; Ghosh, S.K. New scale of atomic orbital radii and its relationships with polarizability, electronegativity, other atomic properties, and bond energies of diatomic molecules. J. Phys. Chem. 1996, 100, 17429–17433. [Google Scholar] [CrossRef]
- Robles, J.; Bartolotti, L.J. Electronegativities, electron affinities, ionization potentials, and hardnesses of the elements within spin polarized density functional theory. J. Am. Chem. Soc. 1984, 106, 3723–3727. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Duley, S. Electron affinity, electronegativity, and electrophilicity of atoms and ions. J. Chem. Eng. Data 2010, 55, 1882–1886. [Google Scholar] [CrossRef]
- Dauben, H.J., Jr.; Wilson, J.D.; Laity, J.L. Diamagnetic susceptibility exaltation as a criterion of aromaticity. J. Am. Chem. Soc. 1968, 90, 811–813. [Google Scholar] [CrossRef]
- Berkowitz, M.; Parr, R.G. Molecular hardness and softness, local hardness and softness, hardness and softness kernels, and relations among these quantities. J. Chem. Phys. 1988, 88, 2554–2557. [Google Scholar] [CrossRef]
- Putz, M.V. Electronegativity and chemical hardness: Different patterns in quantum chemistry. Curr. Phys. Chem. 2011, 1, 111–139. [Google Scholar] [CrossRef]
- Putz, M.V. Compactness aromaticity of atoms in molecules. Int. J. Mol. Sci. 2010, 11, 1269–1310. [Google Scholar] [CrossRef] [Green Version]
- Putz, M.V. Absolute and Chemical Electronegativity and Hardness; NOVA Science Publishers Inc.: New York, NY, USA, 2008. [Google Scholar]
- Putz, M.V. Semiclassical electronegativity and chemical hardness. J. Theor. Comp. Chem. 2007, 6, 33–47. [Google Scholar] [CrossRef]
- Putz, M.V.; Russo, N.; Sicilia, E. Atomic radii scale and related size properties from density functional electronegativity formulation. J. Phys. Chem. A 2003, 107, 5461–5465. [Google Scholar] [CrossRef]
- Putz, M.V. Nanoroots of Quantum Chemistry: Atomic Radii, Periodic Behavior, and Bondons. In Nanoscience and Advancing Computational Methods in Chemistry: Research Progress; Castro, E.A., Haghi, A.K., Eds.; IGI Global: Hershey, PA, USA, 2012; Chapter 4; pp. 103–143. [Google Scholar]
- Ghosh, D.C.; Islam, N. Whether electronegativity and hardness manifest two different descriptors of the one and the same fundamental property of atoms—A quest. Int. J. Quantum Chem. 2011, 111, 40–51. [Google Scholar] [CrossRef]
- Ghosh, D.C.; Islam, N. Whether there is a hardness equalization principle analogous to the electronegativity equalization principle—A quest. Int. J. Quantum Chem. 2011, 111, 1961–1969. [Google Scholar] [CrossRef]
- Putz, M.V.; Russo, N.; Sicilia, E. On the application of the HSAB principle through the use of improved computational schemes for chemical hardness evaluation. J. Comp. Chem. 2004, 25, 994–1003. [Google Scholar] [CrossRef]
- Putz, M.V. On relationship between electronic sharing in bonding and electronegativity equalization of atoms in molecules. Int. J. Chem. Model. 2011, 3, 371–384. [Google Scholar]
- Ciesielski, A.; Krygowski, T.M.; Cyranski, M.K.; Dobrowolski, M.A.; Balaban, A.T. Are thermodynamic and kinetic stabilities correlated? A topological index of reactivity toward electrophiles used as a criterion of aromaticity of polycyclic benzenoid hydrocarbons. J. Chem. Inf. Model. 2009, 49, 369–376. [Google Scholar] [CrossRef]
- Putz, M.V. Quantum parabolic effects of electronegativity and chemical hardness on carbon π-systems. In Carbon Bonding and Structures: Advances in Physics and Chemistry; Putz, M.V., Ed.; Springer: London, UK, 2011; Chapter 1; pp. 1–32. [Google Scholar]
- Putz, M.V. Chemical orthogonal spaces (COSs): From structure to reactivity to biological activity. Int. J. Chem. Model. 2013, 5, 1–34. [Google Scholar]
- Putz, M.V. Bonding in orthogonal space of a chemical structure: From in cerebro to in silico. Int. J. Chem. Model. 2013, 5, 369–396. [Google Scholar]
- Putz, M.V.; Ori, O.; Cataldo, F.; Putz, A.M. Parabolic reactivity “coloring” molecular topology: Application to carcinogenic PAHs. Curr. Org. Chem. 2013, 17, 2816–2830. [Google Scholar] [CrossRef]
- Tudoran, M.A.; Putz, M.V. Molecular graph theory: From adjacency information to colored topology by chemical reactivity. Curr. Org. Chem. 2015, 19, 359–386. [Google Scholar] [CrossRef]
- Gutman, I.; Milun, M.; Trinastić, N. Graph theory and molecular orbitals. 19. Nonparametric resonance energies of arbitrary conjugated systems. J. Am. Chem. Soc. 1977, 99, 1692–1704. [Google Scholar] [CrossRef]
- Putz, M.V.; Tudoran, M.A.; Ori, O. Topological organic chemistry: From distance matrix to Timisoara eccentricity. Curr. Org. Chem. 2015, 19, 249–273. [Google Scholar] [CrossRef]
- Putz, M.V.; Tudoran, M.A.; Mirica, M.C. Quantum dots searching for bondots. Towards sustainable sensitized solar cells. In Sustainable Nanosystems Development, Properties, and Applications; IGI Global: Hershey, PA, USA, 2016; Chapter 9. [Google Scholar]
- Putz, M.V.; Tudoran, M.A.; Mirica, M.C. Bondonic electrochemistry: Basic concepts and sustainable prospects. In Sustainable Nanosystems Development, Properties, and Applications; IGI Global: Hershey, PA, USA, 2016; Chapter 10. [Google Scholar]
- Putz, M.V. Chemical Orthogonal Spaces. In Mathematical Chemistry Monographs; University of Kragujevac: Kragujevac, Serbia, 2012; Volume 14. [Google Scholar]
- Nalewajski, R.F. Electrostatic effects in interaction between hard (soft) acids and bases. J. Am. Chem. Soc. 1984, 106, 944–945. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Kohn-Sham description of equilibria and charge transfer in reactive systems. Int. J. Quantum Chem. 1998, 69, 591–605. [Google Scholar] [CrossRef]
- Putz, M.V.; Putz, A.M. DFT chemical reactivity driven by biological activity: Applications for the toxicological fate of chlorinated PAHs. Struct. Bond. 2013, 150, 181–232. [Google Scholar]
- Putz, M.V.; Dudaș, N.A. Variational principles for mechanistic quantitative structure–activity relationship (QSAR) studies: Application on uracil derivatives’ anti-HIV action. Struct. Chem. 2013, 24, 1873–1893. [Google Scholar] [CrossRef]
- Putz, M.V.; Dudaș, N.A. Determining Chemical Reactivity Driving Biological Activity from SMILES Transformations: The Bonding Mechanism of Anti-HIV Pyrimidines. Molecules 2013, 18, 9061–9116. [Google Scholar] [CrossRef] [Green Version]
- Putz, M.V.; Tudoran, M.A.; Putz, A.M. Structure properties and chemical-bio/ecological of pah interactions: From synthesis to cosmic spectral lines, nanochemistry, and lipophilicity-driven reactivity. Curr. Org. Chem. 2013, 17, 2845–2871. [Google Scholar] [CrossRef]
- Kovacevic, N.; Kokalj, A. Analysis of molecular electronic structure of imidazole- and benzimidazole-based inhibitors: A simple recipe for qualitative estimation of chemical hardness. Corros. Sci. 2011, 53, 909–921. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C. A new equation based on ionization energies and electron affinities of atoms for calculating of group electronegativity. Comput. Theor. Chem. 2015, 1052, 42–46. [Google Scholar] [CrossRef]
- Guerra, D.; Contreras, R.; Perez, P.; Fuentealba, P. Hardness and softness kernels, and related indices in the spin polarized version of density functional theory. Chem. Phys. Lett. 2006, 419, 37–43. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. A link between the ionization energy ratios of an atom and its electronegativity and hardness. Chem. Phys. Lett. 2006, 431, 195–198. [Google Scholar] [CrossRef]
- Meneses, L.; Fuentealba, P.; Contreras, R. On the variations of electronic chemical potential and chemical hardness induced by solvent effects. Chem. Phys. Lett. 2006, 433, 54–57. [Google Scholar] [CrossRef]
- Tang, C.; Zhu, W.; Deng, K. The evolutions of the structure stability, vibrational frequency, frontier orbital, and electronegativity of the unconventional exohedral fullerenes C64X4 (X = H, F, Cl, Br, and I): A density functional study. J. Mol. Struct. (THEOCHEM) 2009, 909, 43–48. [Google Scholar] [CrossRef]
- Matar, S.F.; Campet, G.; Subramanian, M.A. Electronic properties of oxides: Chemical and theoretical approaches. Prog. Sol. State Chem. 2011, 39, 70–95. [Google Scholar] [CrossRef]
- Noorizadeh, S. The maximum hardness and minimum polarizability principles in accordance with the Bent rule. J. Mol. Struct. (THEOCHEM) 2005, 713, 27–32. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; Luis, J.M.; Duran, M.; Sola, M. An assessment of a simple hardness kernel approximation for the calculation of the global hardness in a series of Lewis acids and bases. J. Mol. Struct. (THEOCHEM) 2005, 727, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Putz, M.V. Valence atom with Bohmian quantum potential: The golden ratio approach. Chem. Central J. 2012, 6, 135. [Google Scholar] [CrossRef] [Green Version]
- Nataraj, A.; Balachandran, V.; Karthick, T. Molecular orbital studies (hardness, chemical potential, electrophilicity, and first electron excitation), vibrational investigation and theoretical NBO analysis of 2-hydroxy-5-bromobenzaldehyde by density functional method. J. Mol. Struct. 2013, 1031, 221–233. [Google Scholar] [CrossRef]
- Demircioglu, Z.; Kastas, C.A.; Buyukgungor, O. Theoretical analysis (NBO, NPA, Mulliken Population Method) and molecular orbital studies (hardness, chemical potential, electrophilicity and Fukui function analysis) of (E)-2-((4-hydroxy-2-methylphenylimino)methyl)-3-methoxyphenol. J. Mol. Struct. 2015, 1091, 183–195. [Google Scholar] [CrossRef]
- Yıldız, M.; Karpuz, O.; Zeyrek, C.T.; Boyacıoglu, B.; Dal, H.; Demir, N.; Yıldırım, N.; Unver, H. Synthesis, biological activity, DNA binding and anion sensors, molecular structure and quantum chemical studies of a novel bidentate Schiff base derived from 3,5-bis(triflouromethyl)aniline and salicylaldehyde. J. Mol. Struct. 2015, 1094, 148–160. [Google Scholar] [CrossRef]
- Schroder, D.; Schroeter, K.; Zummack, W.; Schwarz, H. Charge inversion as a structural probe for C6H5+ and C6H6+. ations. J. Am. Soc. Mass. Spectrom. 1999, 10, 878–882. [Google Scholar] [CrossRef] [Green Version]
- Kokalj, A. On the HSAB based estimate of charge transfer between adsorbates and metal surfaces. Chem. Phys. 2012, 393, 1–12. [Google Scholar] [CrossRef]
- Lide, D.R.; Frederikse, H.P.R. (Eds.) CRC Handbook of Chemistry and Physics 1996–1997, 77th ed.; CRC Press: Boca Raton, FL, USA, 1996; Section 5; pp. 1–60. [Google Scholar]
- Energy Units Converter. Available online: http://www.colby.edu/chemistry/PChem/Hartree.html (accessed on 26 December 2020).
- Light, K.J.; Allison, J. Mechanistic considerations of the protonation and fragmentation of highly functionalized molecules in fast atom bombardment: High resolution mass spectrometry and tandem mass spectrometry analysis of the ions formed by fast atom bombardment of digoxin and related cardiac glycosides. J. Am. Soc. Mass. Spectrom. 1990, 1, 455–472. [Google Scholar]
- Bakhtiar, R.; Jacobson, D.B. Transition-metal mediated heteroatom removal by reactions of FeL+ [L=O, C4H6,c-C5H6, c-C5H5, C6H6, C5H4(=CH2)] with furan, thiophene, and pyrrole in the gas phase. J. Am. Soc. Mass. Spectrom. 1996, 7, 938–952. [Google Scholar] [CrossRef] [Green Version]
- Sobocinski, P.; Pesic, Z.D.; Hellhammer, R.; Stolterfoht, N.; Chesnel, J.-Y.; Legendre, S.; Sulik, B. Fragmentation of H2O molecules following interaction with slow He2+ ions. Nucl. Instr. Meth. Phys. Res. B 2005, 233, 207–212. [Google Scholar] [CrossRef]
- Moreh, R.; Finkelstein, Y.; Vos, M. Electron scattering as a tool to study zero-point kinetic energies of atoms in molecules. Nucl. Instr. Meth. Phys. Res. B 2015, 354, 37–41. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, M.Y.; Kim, E.S.; Han, H.S.; Seo, G. H2-SCR of NO on Pt–MnOx catalysts: Reaction path via NH3 formation. Appl. Catal. A Gen. 2011, 395, 120–128. [Google Scholar] [CrossRef]
- Donazzi, A.; Livio, D.; Diehm, C.; Beretta, A.; Groppi, G.; Forzatti, P. Effect of pressure in the autothermal catalytic partial oxidation of CH4 and C3H8: Spatially resolved temperature and composition profiles. Appl. Catal. A Gen. 2014, 469, 52–64. [Google Scholar] [CrossRef]
- Kobzarenko, A.V.; Sukhov, F.F.; Orlov, A.Y.; Kovalev, G.V.; Baranova, I.A.; Feldman, V.I. Effect of molecular structure on fragmentation of isolated organic molecules in solid rare gas matrices. Rad. Phys. Chem. 2012, 81, 1434–1439. [Google Scholar] [CrossRef]
- Sulkes, M. Selective bond fragmentation in pulsed laser ablation of ring strained molecules: Evidence for a thermal mechanism. Chem. Phys. Lett. 2005, 402, 165–169. [Google Scholar] [CrossRef]
- Pillai, E.D.; Molek, K.S.; Duncan, M.A. Growth and photodissociation of U+(C6H6)n (n = 1–3) and UO+m(C6H6) (m = 1,2) complexes. Chem. Phys. Lett. 2005, 405, 247–251. [Google Scholar] [CrossRef]
- Sadr-Arani, L.; Mignon, P.; Chermette, H.; Douki, T. Theoretical and experimental study of the fragmentation of protonated uracil. Chem. Phys. Lett. 2014, 605–606, 108–114. [Google Scholar] [CrossRef]
- Hufsky, F.; Scheubert, K.; Böcker, S. Computational mass spectrometry for small-molecule fragmentation. Trend. Anal. Chem. 2014, 53, 41–48. [Google Scholar] [CrossRef]
- Green, F.M.; Gilmore, I.S.; Seah, M.P. G-SIMS and SMILES: Simulated fragmentation pathways for identification of complex molecules, amino acids and peptides. Appl. Surf. Sci. 2008, 255, 852–855. [Google Scholar] [CrossRef]
- Green, F.M.; Dell, E.J.; Gilmore, I.S.; Seah, M.P. Identification of complex molecules at surfaces: G-SIMS and SMILES fragmentation pathways. Int. J. Mass Spectrom. 2008, 272, 38–47. [Google Scholar] [CrossRef]
- Rahemi, N.; Haghighi, M.; Babaluo, A.A.; Allahyari, S.; Jafari, M.F. Syngas production from reforming of greenhouse gases CH4/CO2 over Ni–Cu/Al2O3 nanocatalyst: Impregnated vs. plasma-treated catalyst. Energ. Convers. Manag. 2014, 84, 50–59. [Google Scholar] [CrossRef]
- Cimpoesu, F.; Zaharia, A.; Stamate, D.; Panait, P.; Oprea, C.I.; Girtu, M.A.; Ferbinteanu, M. New insights in the bonding regime and ligand field in Wernerian complexes. A density functional study. Polyhedron 2013, 52, 183–195. [Google Scholar] [CrossRef]
- Valencia, I.; Tavizón, G.; Barba-Behrens, N.; Castro, M. Rice-ball structures of iron–benzene clusters, Fe4–C6H6)m, m ≤ 3. A density functional study. Chem. Phys. 2011, 390, 51–59. [Google Scholar] [CrossRef]
- Tamuliene, J.; Romanova, L.G.; Vukstich, V.S.; Snegursky, A.V. Mechanisms of the electron-impact-induced glycine molecule fragmentation. Chem. Phys. 2012, 404, 36–41. [Google Scholar] [CrossRef]
- Tamuliene, J.; Romanova, L.G.; Vukstich, V.S.; Snegursky, A.V. Mechanisms of the electron-impact-induced methionine molecule fragmentation. Chem. Phys. 2012, 404, 74–81. [Google Scholar] [CrossRef]
- Georgiev, S.; Neusser, H.J. Mass analyzed threshold ionization of hydrogen bonded clusters of biological molecules: The 3-methylindole·C6H6 complex. J. Electron. Spectrosc. Relat. Phenom. 2005, 142, 207–213. [Google Scholar] [CrossRef]
- Mitsuke, K. Photofragmentaion mechanisms of H2O studied by ultraviolet dispersed spectroscopy. J. Electron. Spectrosc. Relat. Phenom. 2005, 144–147, 131–133. [Google Scholar] [CrossRef]
- Matito, E.; Putz, M.V. New Link between Conceptual Density Functional Theory and Electron Delocalization. J. Phys. Chem. A 2011, 115, 12459–12462. [Google Scholar] [CrossRef]
- Putz, M.V.; Chattaraj, P.K. Electrophilicity Kernel and its Hierarchy through Softness in Conceptual Density Functional Theory. Int. J. Quantum Chem. 2013, 113, 2163–2171. [Google Scholar] [CrossRef]
- Putz, M.V. Chemical Field Theory: The Inverse Density Problem of Electronegativity and Chemical Hardness for Chemical Bond. Curr. Phys. Chem. 2017, 7, 133–146. [Google Scholar] [CrossRef]
- Gerratt, J.; Cooper, D.L.; Karadakov, P.B.; Raimondi, M. Modern Valence Bond Theory. Chem. Soc. Rev. 1997, 26, 87–100. [Google Scholar] [CrossRef]
- Shaik, S.; Hiberty, P.C. Valence Bond theory, its History, Fundamentals and Applications. A Primer. Rev. Comput. Chem. 2004, 20, 1–100. [Google Scholar]
- Glendening, E.D.; Weinhold, F. Natural Resonance Theory: I. General Formalism. J. Comput. Chem. 1998, 19, 593–609. [Google Scholar] [CrossRef]
- Glendening, E.D.; Weinhold, F. Natural Resonance Theory: II. Natural Bond Order and Valency. J. Comp. Chem. 1998, 19, 610–627. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Weinhold, F. Natural Resonance Theory: III. Chemical Applications. J. Comp. Chem. 1998, 19, 628–646. [Google Scholar] [CrossRef]
- Putz, M.V.; Russo, N.; Sicilia, E. About the Mulliken Electronegativity in DFT. Theor. Chem. Acc. 2005, 114, 38–45. [Google Scholar] [CrossRef]
- Bader, R.F.W. The zero-flux surface and the topological and quantum definitions of an atom in a molecule. Theor. Chem. Acc. 2001, 105, 276–283. [Google Scholar] [CrossRef]
- Bader, R.F.W. A comment on “Some fundamental problems with zero-flux partitioning of electron densities”. Theor. Chem. Acc. 2002, 107, 381–382; c) Bader, R.F.W. Bond paths are not chemical bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. Hydrogen–Hydrogen Bonding in Planar Biphenyl, Predicted by Atoms-In-Molecules Theory, Does Not Exist. Chem. Eur. J. 2006, 12, 2889–2895. [Google Scholar] [CrossRef]
- Berrah, N.; Fang, L.; Osipov, T.; Murphy, B.; Bostedt, C.; Bozek, J.D. Multiphoton ionization and fragmentation of molecules with the LCLSX-ray FEL. J. Electron. Spectrosc. Relat. Phenom. 2014, 196, 34–37. [Google Scholar] [CrossRef]
- Peng, L.-Y.; Jiang, W.-C.; Geng, J.-W.; Xiong, W.-H.; Gong, Q. Tracing and controlling electronic dynamics in atoms and molecules by attosecond pulses. Phys. Rep. 2015, 575, 1–71. [Google Scholar] [CrossRef]
- Czasch, A.; Schmidt, L.P.H.; Jahnke, T.; Weber, T.; Jagutzki, O.; Schössler, S.; Schöffler, M.S.; Dörner, R.; Schmidt-Böcking, H. Photo induced multiple fragmentation of atoms and molecules: Dynamics of Coulombic many-particle systems studied with the COLTRIMS reaction microscope. Phys. Lett. A 2005, 347, 95–102. [Google Scholar] [CrossRef]
- Muthu, S.; Renuga, S. Molecular orbital studies (hardness, chemical potential, electronegativity and electrophilicity), vibrational spectroscopic investigation and normal coordinate analysis of 5-{1-hydroxy-2-[(propan-2-yl)amino]ethyl}benzene-1,3-diol. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 118, 683–694. [Google Scholar] [CrossRef]
- Verstraelen, T.; Bultinck, P. Can the electronegativity equalization method predict spectroscopic properties? Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.M.; Lin, T.H.; Chen, Y.S.; Chang, C.W.; Huang, K.L.; Wu, F.W.; Hsu, W.J.; Yu, M.P.; Lin, C.; Wang, M.K. A quantum chemical approach using classical concepts to characterization and descriptive analysis of various reactions of metal ions and organic compounds. Chemometr. Intell. Lab. Syst. 2014, 136, 155–163. [Google Scholar] [CrossRef]
- Smith, D.W. A new approach to the relationship between bond energy and electronegativity. Polyhedron 2007, 26, 519–523. [Google Scholar] [CrossRef]
- Li, K.; Yang, P.; Xue, D. Anisotropic hardness prediction of crystalline hard materials from the electronegativity. Acta Mater. 2012, 60, 35–42. [Google Scholar] [CrossRef]
- Gatchell, M.; Stockett, M.H.; Rousseau, P.; Chen, T.; Kulyk, K.; Schmidt, H.T.; Chesnel, J.Y.; Domaracka, A.; Méry, A.; Maclot, S.; et al. Non-statistical fragmentation of PAHs and fullerenes in collisionswith atoms. Int. J. Mass Spectrom. 2014, 365–366, 260–265. [Google Scholar] [CrossRef]
- Laskin, J.; Futrell, J.H. New approach for studying slow fragmentation kinetics in FT-ICR: Surface-induced dissociation combined with resonant ejection. Int. J. Mass Spectrom. 2015, 378, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.C., II; Chuang, S.S.C. Investigating the CH4 reaction pathway on a novel LSCF anode catalyst in the SOFC. Catal. Commun. 2009, 10, 772–776. [Google Scholar] [CrossRef]
- Whitney, C.K. Closing in on chemical bonds by opening up relativity theory. Int. J. Mol. Sci. 2008, 9, 272–298. [Google Scholar] [CrossRef] [Green Version]
- Rahm, M.; Hoffmann, R. Toward an experimental quantum chemistry: Exploring a new energy partitioning. J. Am. Chem. Soc. 2015, 137, 10282–10291. [Google Scholar] [CrossRef]
- Von Szentpály, L. Symmetry laws improve electronegativity equalization by orders of magnitude and call for a paradigm shift in conceptual density functional theory. J. Phys. Chem. A 2015, 119, 1715–1722. [Google Scholar] [CrossRef]
- Moens, J.; Geerlings, P.; Roos, G. A conceptual DFT approach for the evaluation and interpretation of redox potentials. Chem. Eur. J. 2007, 13, 8174–8184. [Google Scholar] [CrossRef] [PubMed]
- Moens, J.; Jaque Olmedo Pablo, C.; De Proft, F.; Geerlings, P. The Study of Redoxreactions based on Conceptual DFT Principles: EEM and Vertical Quantities. J. Phys. Chem. A 2008, 112, 6023–6031. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Maynard, A.T.; Huang, M.; Rice, W.G.; Covell, D.G. Reactivity of the HIV-1 nucleocapsid protein p7 zinc finger domains from the perspective of density-functional theory. Proc. Natl. Acad. Sci. USA 1998, 95, 11578–11583. [Google Scholar] [CrossRef] [Green Version]
- Nalewajski, R.F. Understanding Electronic Structure and Chemical Reactivity: Quantum-Information Perspective. Appl. Sci. 2019, 9, 1262. [Google Scholar] [CrossRef] [Green Version]
- Nalewajski, R.F. Phase Equalization, Charge Transfer, Information Flows and Electron Communications in Donor–Acceptor Systems. Appl. Sci. 2020, 10, 3615. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Type | Parr–Pearson | COS | |
|---|---|---|---|
| AB | |||
| AA | |||
| AB | ΔfH (eV) | a | a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PP | COS | PP | COS | PP | COS | PP | COS | PP | COS | ||||||
| HF | −2.833 | 6.42 | 7.01 | 7.17 | 10.41 | 0.121 | 0.184 | 6.70 | 6.68 | 8.71 | 8.58 | −0.195 | −0.676 | 0.65 | 0.643 |
| IF | −0.992 | 3.69 | 7.01 | 6.76 | 10.41 | 0.171 | −0.173 | 4.83 | 4.95 | 8.02 | 8.15 | −0.311 | 0.781 | 0.829 | 0.823 |
| BrF | −0.972 | 4.22 | 7.01 | 7.59 | 10.41 | 0.126 | −0.157 | 5.27 | 5.36 | 8.65 | 8.74 | −0.177 | 0.751 | 0.821 | 0.815 |
| HCl | −0.957 | 6.42 | 4.68 | 7.17 | 8.30 | 0.051 | 0.328 | 5.41 | 5.55 | 7.82 | 7.74 | −0.029 | −0.971 | 0.723 | 0.698 |
| ClF | −0.521 | 4.68 | 7.01 | 8.30 | 10.41 | 0.09 | −0.144 | 5.61 | 5.69 | 9.14 | 9.21 | −0.01 | 0.723 | 0.815 | 0.810 |
| HBr | −0.376 | 6.42 | 4.22 | 7.17 | 7.59 | 0.02 | 0.341 | 5.09 | 5.26 | 7.42 | 7.39 | −0.004 | −0.954 | 0.729 | 0.702 |
| BrCl | 0.151 | 4.22 | 4.68 | 7.59 | 8.30 | 0.04 | −0.013 | 4.44 | 4.44 | 7.93 | 7.93 | −0.014 | 0.05 | 0.893 | 0.893 |
| ICl | 0.184 | 3.69 | 4.68 | 6.76 | 8.30 | 0.092 | −0.029 | 4.13 | 4.13 | 7.44 | 7.45 | −0.071 | 0.111 | 0.901 | 0.901 |
| HI | 0.275 | 6.42 | 3.69 | 7.17 | 6.76 | −0.02 | 0.358 | 4.69 | 4.9 | 6.91 | 6.94 | −0.004 | −0.928 | 0.737 | 0.708 |
| IBr | 0.423 | 3.69 | 4.22 | 6.76 | 7.59 | 0.052 | −0.017 | 3.94 | 3.94 | 7.15 | 7.15 | −0.022 | 0.061 | 0.908 | 0.908 |
| AB | ΔfHa (eV) | b | b | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PP | COS | PP | COS | PP | COS | PP | COS | PP | P | ||||||
| LiF | −6.384 | 2.39 | 7.01 | 3.01 | 10.41 | 0.394 | 0.113 | 3.56 | 3.47 | 4.89 | 4.74 | −1.456 | −0.246 | 0.686 | 0.683 |
| NaF | −5.976 | 2.30 | 7.01 | 2.85 | 10.41 | 0.406 | 0.123 | 3.46 | 3.36 | 4.72 | 4.56 | −1.535 | −0.255 | 0.681 | 0.677 |
| NaCl | −4.262 | 2.30 | 4.68 | 2.85 | 8.30 | 0.39 | 0.267 | 3.08 | 2.96 | 4.65 | 4.36 | −1.064 | −0.477 | 0.753 | 0.737 |
| LiCl | −4.235 | 2.39 | 4.68 | 3.01 | 8.30 | 0.374 | 0.257 | 3.16 | 3.05 | 4.8 | 4.53 | −0.99 | −0.482 | 0.758 | 0.743 |
| NaBr | −3.743 | 2.30 | 4.22 | 2.85 | 7.59 | 0.363 | 0.28 | 2.98 | 2.87 | 4.52 | 4.26 | −0.861 | −0.483 | 0.759 | 0.742 |
| LiBr | −3.640 | 2.39 | 4.22 | 3.01 | 7.59 | 0.346 | 0.27 | 3.05 | 2.95 | 4.67 | 4.42 | −0.793 | −0.488 | 0.764 | 0.748 |
| NaI | −2.983 | 2.30 | 3.69 | 2.85 | 6.76 | 0.326 | 0.296 | 2.83 | 2.75 | 4.35 | 4.11 | −0.638 | −0.488 | 0.768 | 0.748 |
| LiI | −2.803 | 2.39 | 3.69 | 3.01 | 6.76 | 0.308 | 0.286 | 2.9 | 2.82 | 4.48 | 4.26 | −0.578 | −0.494 | 0.773 | 0.754 |
| LiH | −0.938 | 2.39 | 6.42 | 3.01 | 7.17 | 0.236 | −0.071 | 3.48 | 3.54 | 4.14 | 4.2 | −0.491 | 0.159 | 0.594 | 0.593 |
| NaH | −0.584 | 2.30 | 6.42 | 2.85 | 7.17 | 0.248 | −0.061 | 3.39 | 3.44 | 3.99 | 4.04 | −0.535 | 0.13 | 0.589 | 0.588 |
| A2 | S (J∙K−1∙mol−1) a,b (eV) | ΔfH b (eV) | ΔΔH b (eV) | Indices b (eV) | ΔNAA | ηAA (eV) | χAA (eV) | EAA (eV) | Cπ(AA) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ηA | χA | PP | COS | PP | COS | PP | COS | PP | COS | PP | COS | ||||
| Cl2 (g) | 0.689 | 0 | 0.095 | 4.68 | 8.30 | 0 | 0.228 | 4.68 | 5.25 | 8.30 | 7.98 | 0 | −0.774 | 0.887 | 0.761 |
| Br2 (g) | 0.756 | 0.32 | 0.101 | 4.22 | 7.59 | 0 | 0.237 | 4.22 | 4.75 | 7.59 | 7.3 | 0 | −0.73 | 0.889 | 0.768 |
| I2 (g) | 0.806 | 0.647 | 0.105 | 3.69 | 6.76 | 0 | 0.248 | 3.69 | 4.17 | 6.76 | 6.5 | 0 | −0.677 | 0.916 | 0.778 |
| F2 (g) | 0.627 | 0 | 0.091 | 7.01 | 10.41 | 0 | 0.135 | 7.01 | 7.51 | 10.41 | 10.07 | 0 | −0.613 | 0.743 | 0.671 |
| O2 (g) | 0.634 | 0 | 0.0890 | 6.08 | 7.53 | 0 | 0.063 | 6.08 | 6.28 | 7.53 | 7.37 | 0 | −0.22 | 0.619 | 0.587 |
| H2 (g) | 0.404 | 0 | 0.088 | 6.42 | 7.17 | 0 | 0.03 | 6.42 | 6.52 | 7.17 | 7.08 | 0 | −0.103 | 0.558 | 0.543 |
| N2 (g) | 0.592 | 0 | 0.0899 | 7.23 | 7.27 | 0 | 0.001 | 7.23 | 7.235 | 7.27 | 7.265 | 0 | −0.005 | 0.503 | 0.502 |
| Paths | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H + O | 0.014 | 0.061 | 6.25 | 6.25 | 7.35 | 7.35 | −0.003 | −0.212 | 0.589 | 0.588 |
| 2 | (H + O)COS + H | −0.007 | 0.03 | 6.33 | 6.34 | 7.26 | 7.26 | −6.4 × 10−4 | 0.11 | 0.573 | 0.573 |
| 3 | H2COS + O | 0.018 | 0.076 | 6.29 | 6.3 | 7.31 | 7.3 | −0.004 | −0.26 | 0.581 | 0.58 |
| Paths | |||||||
|---|---|---|---|---|---|---|---|
| 1 | N + H | 6.8 | 6.81 | 7.22 | 7.22 | 0.53 | 0.53 |
| 2 | (N + H)COS + H | 6.61 | 6.61 | 7.19 | 7.19 | 0.544 | 0.544 |
| 3 | N + H2COS | 6.86 | 6.86 | 7.17 | 7.17 | 0.523 | 0.522 |
| 4 | ((N + H)COS + H)COS + H | 6.51 | 6.51 | 7.18 | 7.18 | 0.551 | 0.551 |
| 5 | (N + H)COS + H2COS | 6.66 | 6.66 | 7.15 | 7.15 | 0.536 | 0.536 |
| 6 | (N + H2COS)COS + H | 6.63 | 6.64 | 7.17 | 7.17 | 0.541 | 0.540 |
| No. | Paths | ||||||
|---|---|---|---|---|---|---|---|
| 1 | C + H | 5.62 | 5.65 | 6.66 | 6.68 | 0.593 | 0.592 |
| 2 | (C + H)COS + H | 6.01 | 6.02 | 6.91 | 6.91 | 0.574 | 0.574 |
| 3 | C + H2COS | 5.66 | 5.69 | 6.62 | 6.64 | 0.585 | 0.583 |
| 4 | (C + H)COS + H2COS | 6.05 | 6.06 | 6.87 | 6.87 | 0.567 | 0.566 |
| 5 | (C + H2COS)COS + H | 6.03 | 6.04 | 6.89 | 6.89 | 0.57 | 0.57 |
| 6 | ((C + H)COS + H)COS + H | 6.21 | 6.22 | 7.04 | 7.04 | 0.566 | 0.566 |
| 7 | ((C + H)COS + H)COS+ H2COS | 6.26 | 6.26 | 6.99 | 6.99 | 0.558 | 0.558 |
| 8 | (C + H2COS)COS+ H2COS | 6.07 | 6.08 | 6.85 | 6.85 | 0.563 | 0.562 |
| 9 | ((C + H)COS + H2COS)COS + H | 6.23 | 6.23 | 7.02 | 7.02 | 0.562 | 0.562 |
| 10 | ((C + H2COS)COS + H)COS + H | 6.22 | 6.22 | 7.03 | 7.03 | 0.564 | 0.564 |
| 11 | (((C + H)COS + H)COS + H)COS + H | 6.31 | 6.31 | 7.1 | 7.1 | 0.562 | 0.562 |
| Paths | |||||||
|---|---|---|---|---|---|---|---|
| 1 | CH + CH + CH + CH + CH + CH (+CH) | 5.65 | 5.65 | 6.68 | 6.68 | 0.590 (362) | 0.590 (361) |
| 2 | CH + CH + C2H2 + C2H2 (+CH) | 5.74 (394) | 5.74 (552) | 6.6 | 6.6 | 0.574 (417) | 0.574 (14) |
| 3 | CH + C2H2 + C2H2 + CH (+CH) | 5.68 | 5.68 | 6.66 | 6.66 | 0.586 (456) | 0.586 (435) |
| 4 | CH + C2H2 + CH + C2H2 (+CH) | 5.69 | 5.69 | 6.42 | 6.42 | 0.583 (375) | 0.583 (315) |
| 5 | CH + C2H2 + C3H3 (+CH) | 5.74 (343) | 5.74 (498) | 6.6 | 6.6 | 0.574 (685) | 0.574 (416) |
| 6 | C2H2 + C2H2 + C2H2 (+C2H2) | 5.81 (542) | 5.81 (553) | 6.54 | 6.54 | 0.562 (089) | 0.562 (07) |
| 7 | C2H2 + CH + CH + C2H2 (+C2H2) | 5.77 (245) | 5.77 (25) | 6.58 | 6.58 | 0.571 (047) | 0.571 (026) |
| 8 | C2H2 + C3H3 + CH (+C2H2) | 5.74 (592) | 5.74 (625) | 6.6 | 6.6 | 0.574 (128) | 0.574 (07) |
| 9 | C2H2 + C2H2 + CH + CH (+C2H2) | 5.74 (999) | 5.75 (026) | 6.59 | 6.59 | 0.573 (389) | 0.573 (342) |
| 10 | C2H2 + CH + C3H3 (+C2H2) | 5.81 | 5.82 | 6.54 | 6.54 | 0.562 (357) | 0.562 (34) |
| 11 | C3H3 + C3H3 (+C3H3) | 5.78 | 5.78 | 6.57 | 6.57 | 0.568 (445) | 0.568 (342) |
| 12 | C3H3 + C2H2 + CH (+C3H3) | 5.74 | 5.74 | 6.61 | 6.61 | 0.580 (217) | 0.580 (214) |
| 13 | C3H3 + CH + C2H2 (+C3H3) | 5.73 | 5.73 | 6.61 | 6.61 | 0.577 (135) | 0.577 (133) |
| 14 | C3H3 + CH + CH + CH (+C3H3) | 5.69 | 5.69 | 6.65 | 6.65 | 0.584 (122) | 0.584 (093) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putz, M.V. Chemical Bonding by the Chemical Orthogonal Space of Reactivity. Int. J. Mol. Sci. 2021, 22, 223. https://doi.org/10.3390/ijms22010223
Putz MV. Chemical Bonding by the Chemical Orthogonal Space of Reactivity. International Journal of Molecular Sciences. 2021; 22(1):223. https://doi.org/10.3390/ijms22010223
Chicago/Turabian StylePutz, Mihai V. 2021. "Chemical Bonding by the Chemical Orthogonal Space of Reactivity" International Journal of Molecular Sciences 22, no. 1: 223. https://doi.org/10.3390/ijms22010223
APA StylePutz, M. V. (2021). Chemical Bonding by the Chemical Orthogonal Space of Reactivity. International Journal of Molecular Sciences, 22(1), 223. https://doi.org/10.3390/ijms22010223