Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth

 ,
,  , , ,
, , , 

Abstract
:1. Introduction
2. Results
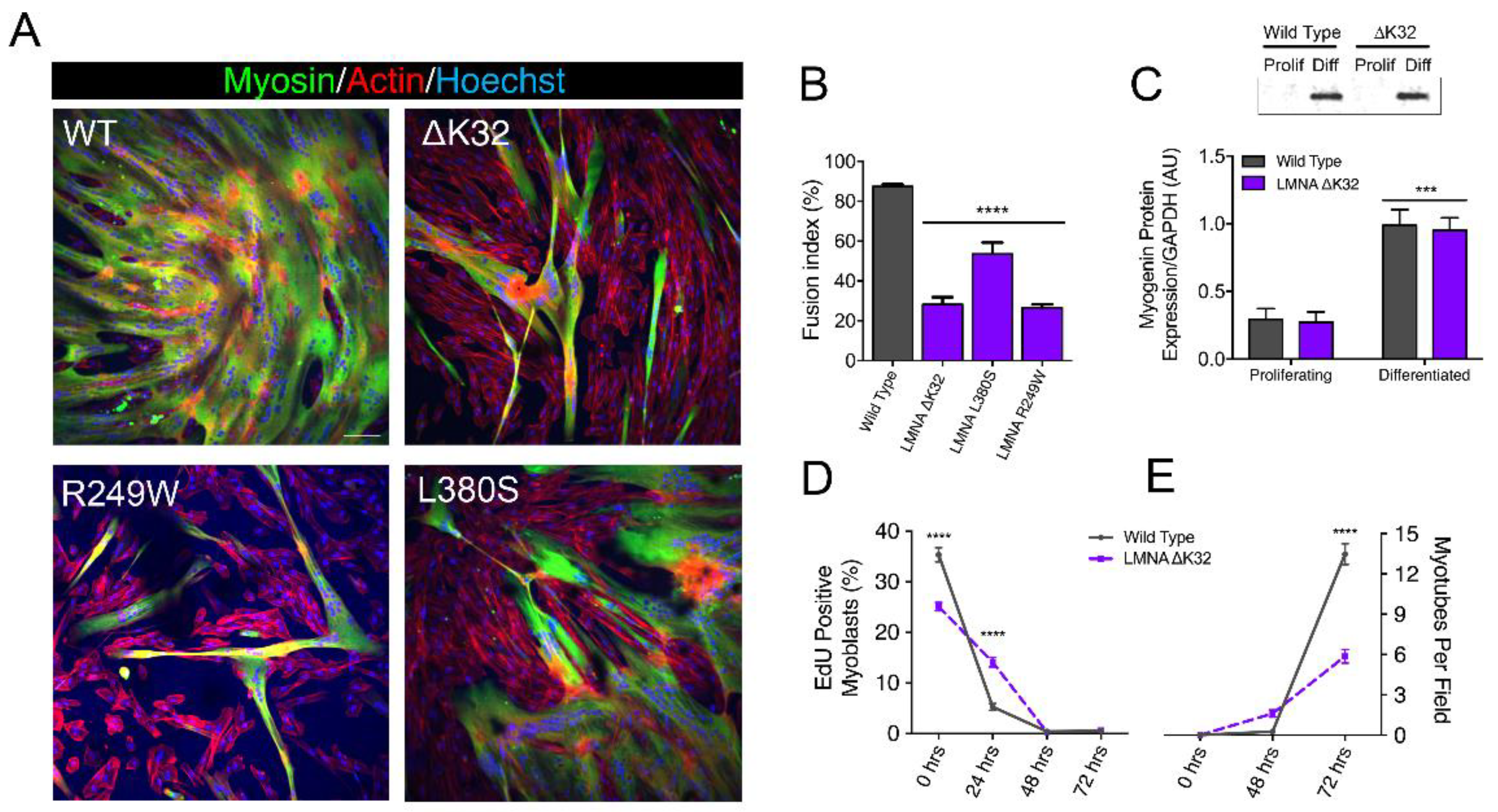
2.1. LMNA-CMD Muscle Stem Cells Exit the Cell Cycle but Exhibit Impaired Fusion
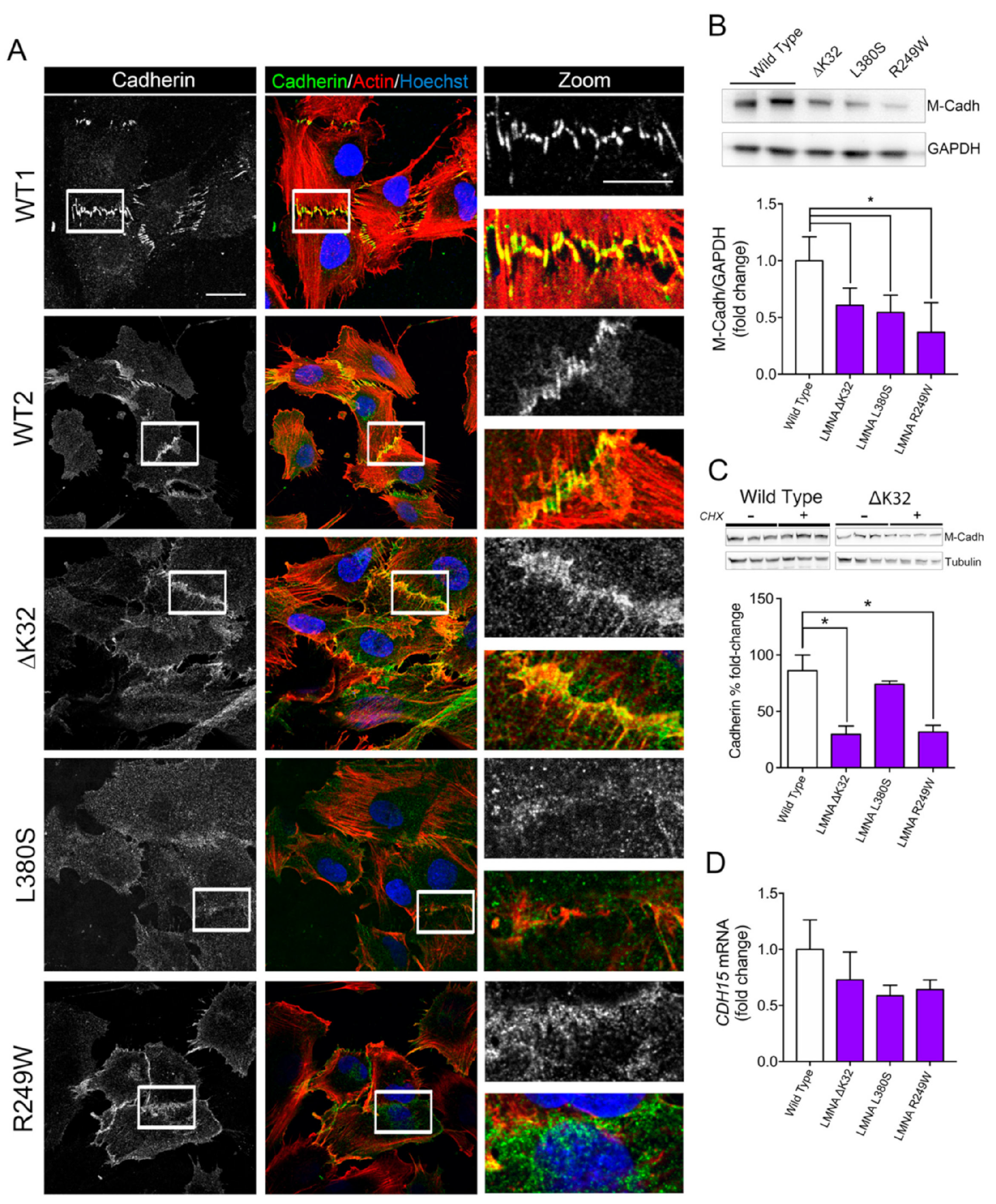
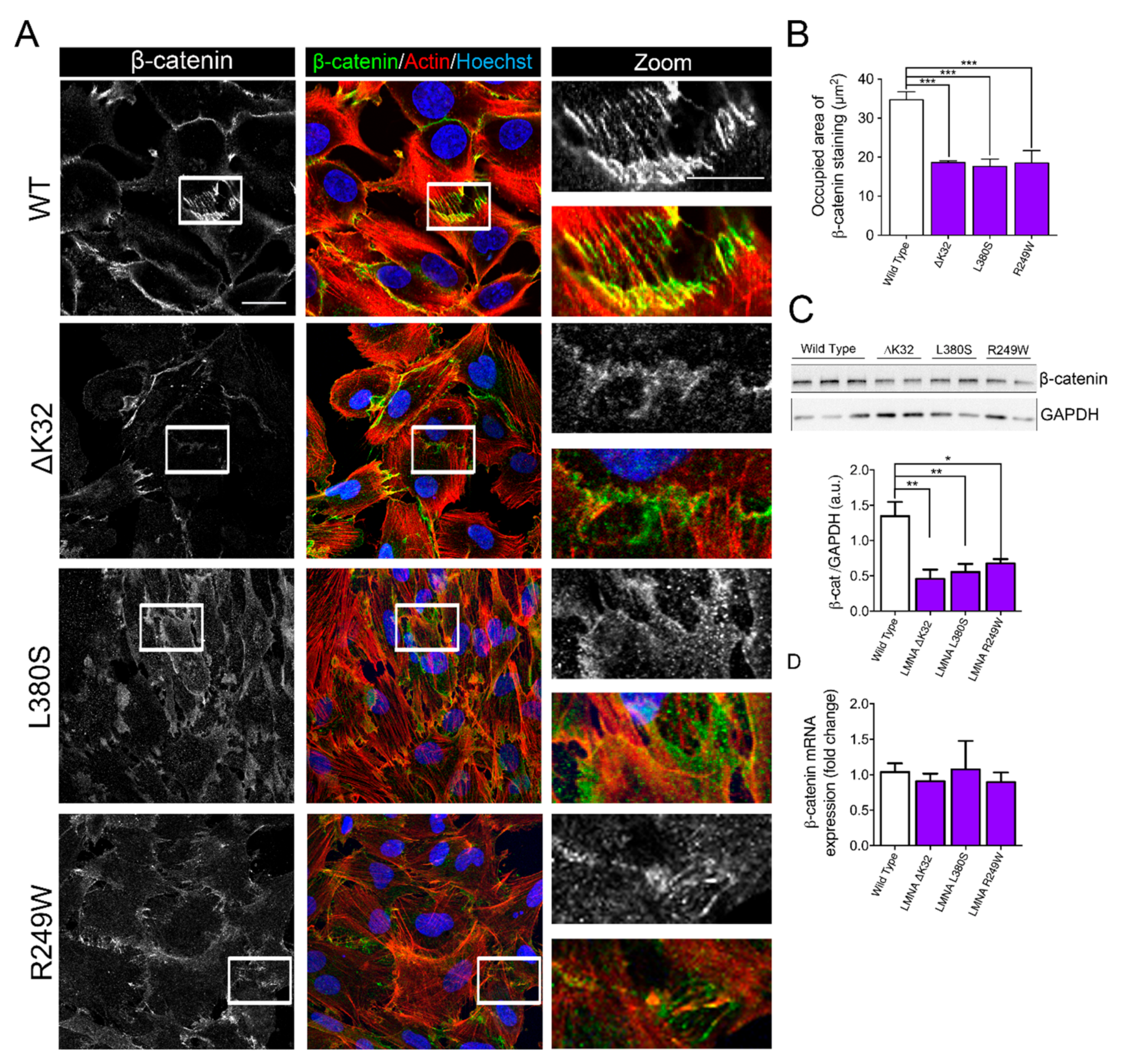
2.2. Impaired Cell–Cell Interactions in LMNA-CMD Mutant Muscle Cells Precursors
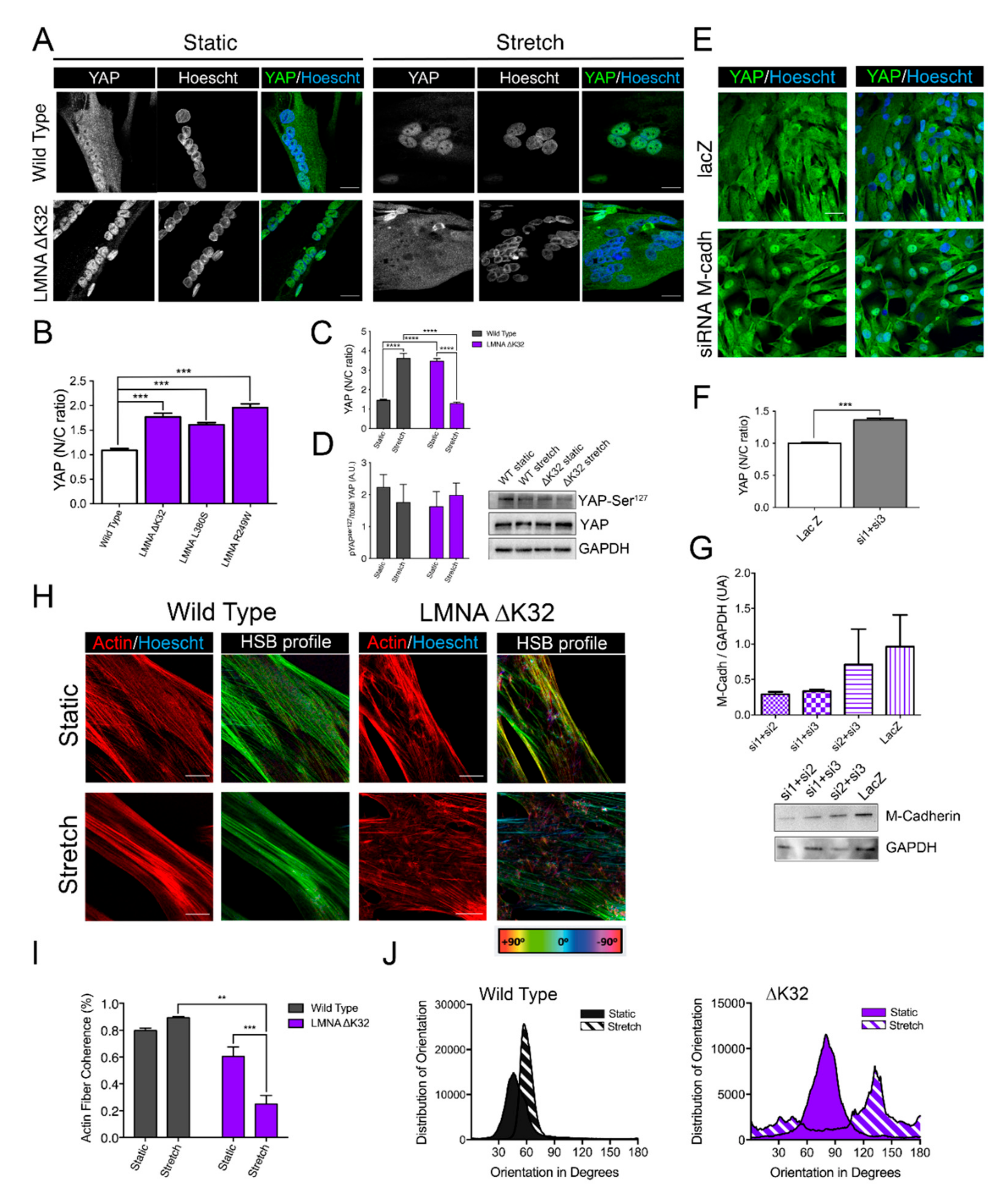
2.3. YAP Nuclear Sequestration in LMNA-CMD Mutant Myotubes
2.4. Adaptability to Mechanical Constraints Is Severely Affected in LMNA-CMD Myotubes
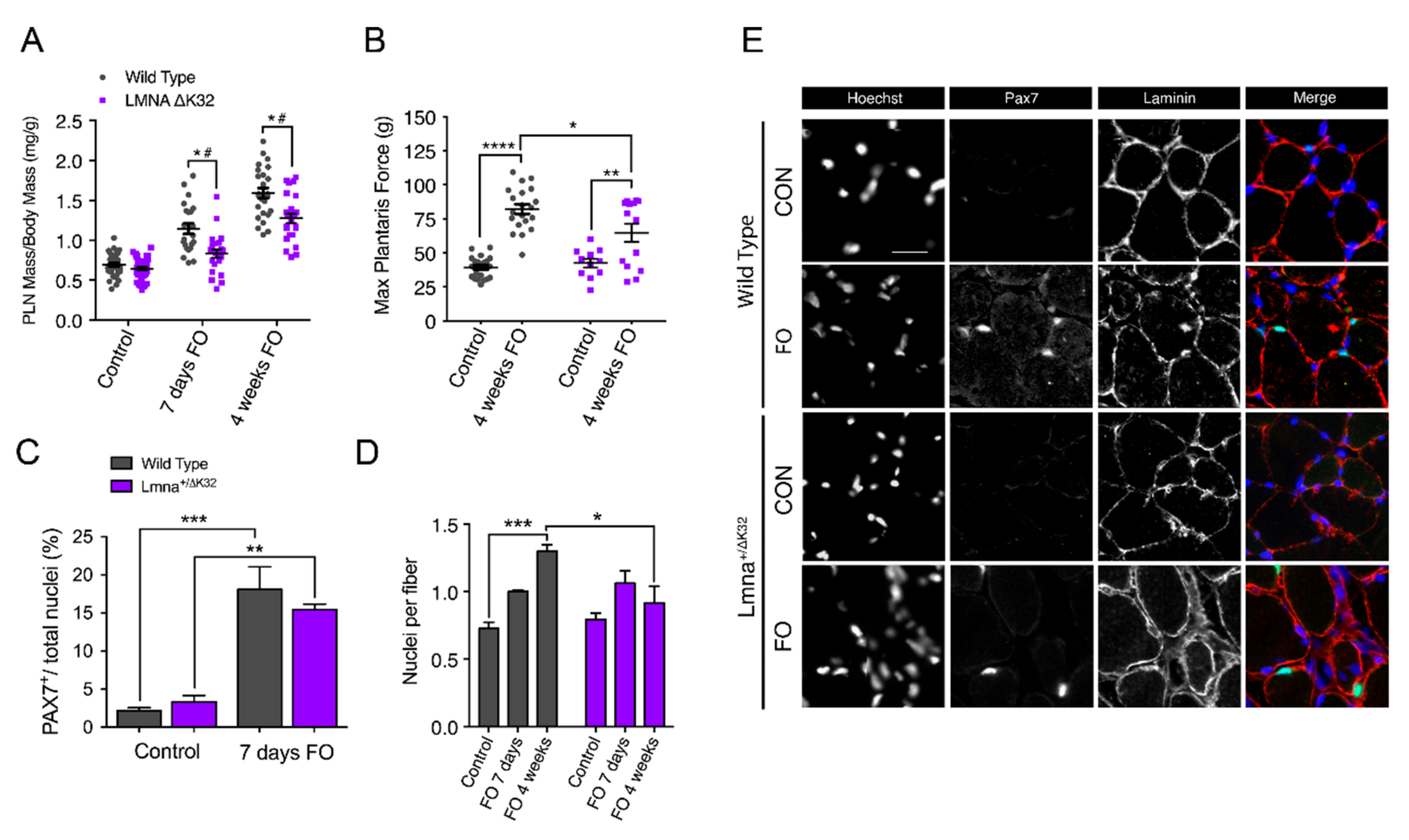
2.5. Defective Muscle Hypertrophy in Lmna ΔK32 Heterozygous Mice
2.6. Defective Myonuclear Accretion in Lmna ΔK32 Heterozygous Mice
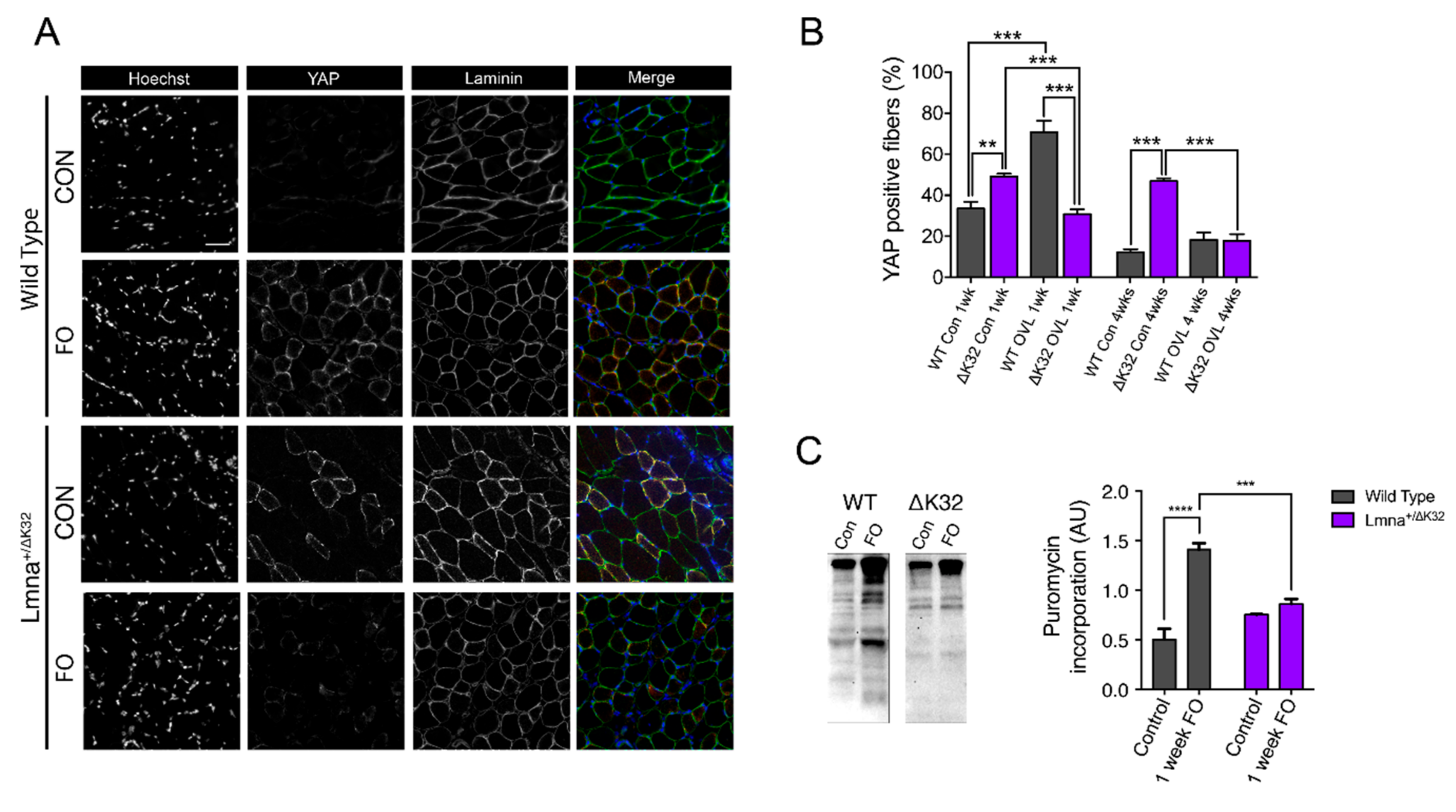
2.7. YAP Abundance Is Higher at Baseline but Decreases after FO in Lmna ΔK32 Heterozygous Mice
2.8. Defective Muscle Protein Synthesis in Lmna ΔK32 Heterozygous Mice
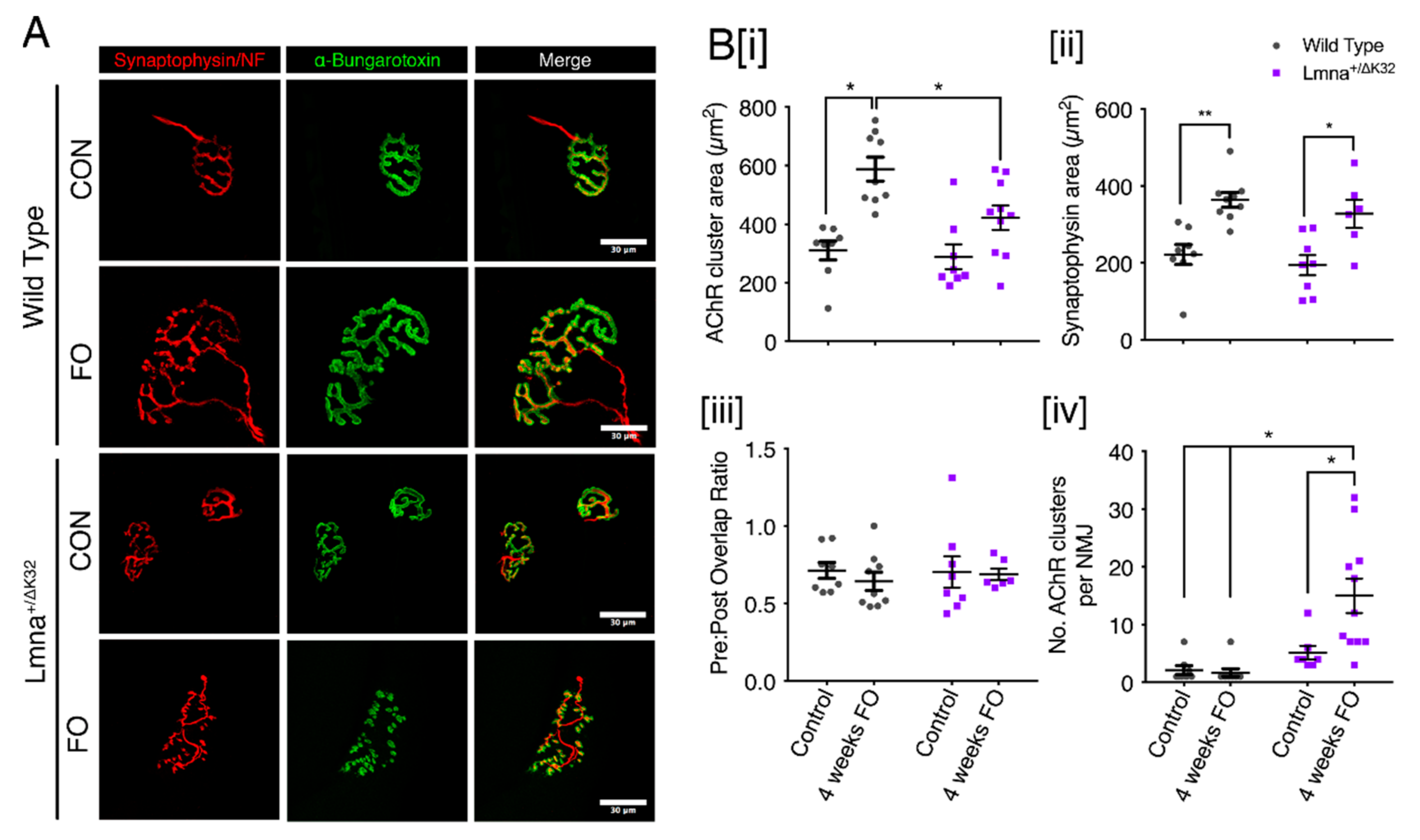
2.9. Neuromuscular Junction Defects after Functional Overload in Lmna ΔK32 Heterozygous Mice
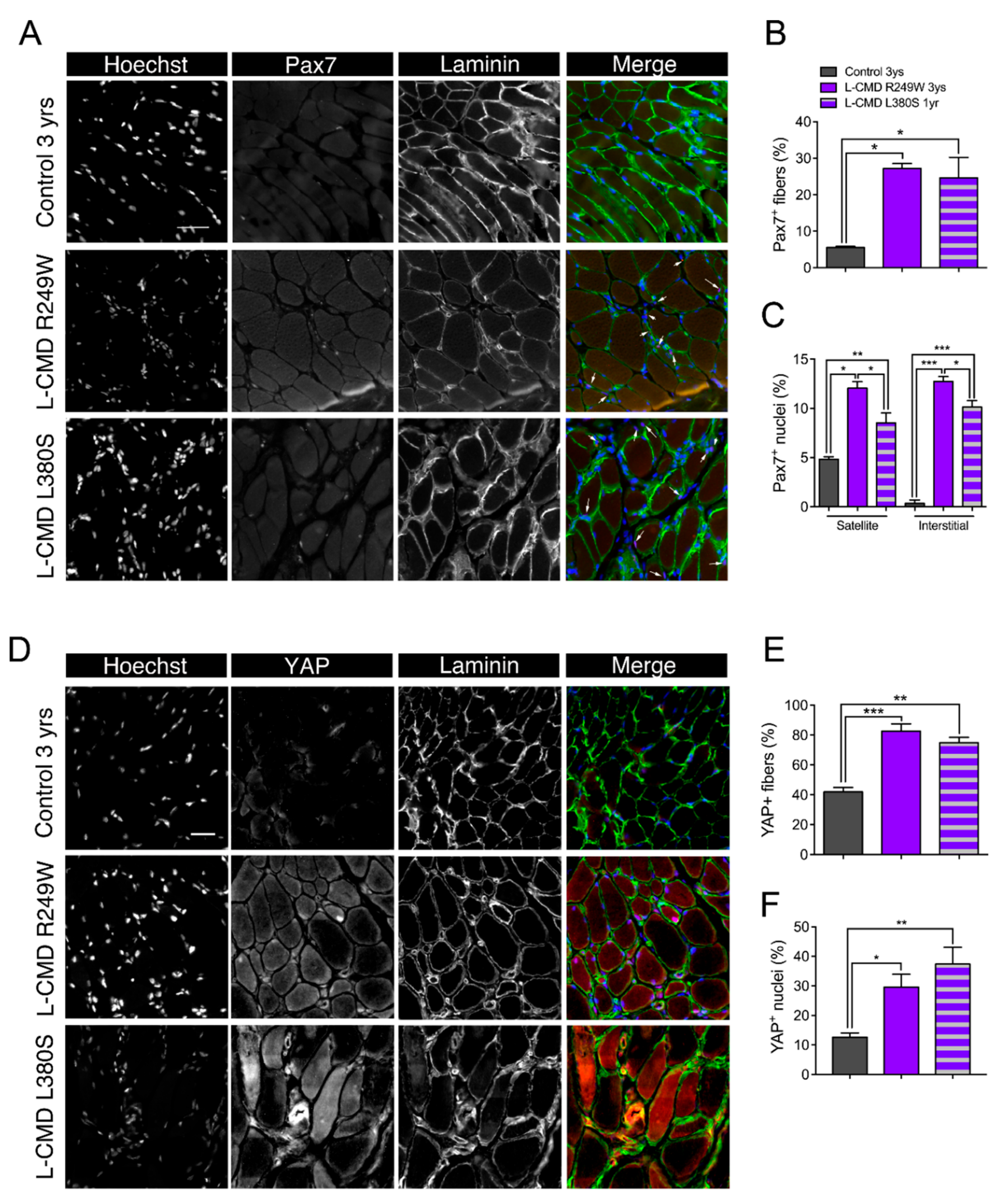
2.10. Muscle Biopsies from LMNA-CMD Patients Revealed Increased Pax7+ Cells and YAP Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.1.1. Human Myoblasts and Cell Culture
4.1.2. Immortalized MyoD-Converted Human Myoblasts
4.1.3. Drug Treatments and siRNA
4.1.4. Cyclic Strain
4.1.5. Immunocytochemistry and Image Analysis
4.1.6. SDS-PAGE and Protein Analysis
4.1.7. Quantification of Gene Expression
4.2. Animal Study
4.2.1. Animals
4.2.2. Functional Overload
4.2.3. In Vivo Estimation of Protein Synthesis
4.2.4. Maximal Force Measures
4.2.5. Immunohistochemistry
4.3. Human Study
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AChR | acetylcholine receptor |
| BSA | bovine serum albumin |
| EDMD | Emery-Dreifuss muscular dystrophy |
| ΔK32 | LMNA c.94_96delAAG, p.Lys32del |
| hEGF | human epidermal growth factor |
| CMD | congenital muscular dystrophy |
| FO | functional overload |
| L380S | LMNA p.Leu380Ser |
| MuSC | muscle stem cell |
| NF | neurofilament |
| PBS | phosphate buffer solution |
| PLN | plantaris muscle |
| R249W | LMNA p.Arg249Trp |
| SDS | sodium dodecylsulfate |
| SRF | serum responsive factor |
| Syn | synaptophysin |
| TBS-T | tris-buffered saline-tween |
| WT | wild-type |
| YAP | yes-associated protein |
References
- Mierzejewski, B.; Archacka, K.; Grabowska, I.; Florkowska, A.; Ciemerych, M.A.; Brzoska, E. Human and mouse skeletal muscle stem and progenitor cells in health and disease. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bershadsky, A.D.; Balaban, N.Q.; Geiger, B. Adhesion-dependent cell mechanosensitivity. Annu. Rev. Cell Dev. Biol. 2003, 19, 677–695. [Google Scholar] [CrossRef] [Green Version]
- Oakes, P.W.; Gardel, M.L. Stressing the limits of focal adhesion mechanosensitivity. Curr. Opin. Cell Biol. 2014, 30, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladoux, B.; Anon, E.; Lambert, M.; Rabodzey, A.; Hersen, P.; Buguin, A.; Silberzan, P.; Mege, R.M. Strength dependence of cadherin-mediated adhesions. Biophys. J. 2010, 98, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Le Duc, Q.; Shi, Q.; Blonk, I.; Sonnenberg, A.; Wang, N.; Leckband, D.; de Rooij, J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 2010, 189, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabdili, H.; Langer, M.; Shi, Q.; Poh, Y.C.; Wang, N.; Leckband, D. Cadherin-dependent mechanotransduction depends on ligand identity but not affinity. J. Cell Sci. 2012, 125 Pt 18, 4362–4371. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, R. Control of satellite cell proliferation. Adv. Exp. Med. Biol. 1990, 280, 147–157. [Google Scholar]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- Schwartz, C.; Fischer, M.; Mamchaoui, K.; Bigot, A.; Lok, T.; Verdier, C.; Duperray, A.; Michel, R.; Holt, I.; Voit, T.; et al. Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci. Rep. 2017, 7, 1253. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Quijano-Roy, S.; Mbieleu, B.; Bonnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2019, 19, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Laine, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127 Pt 13, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, B.A.; Rothballer, A.; Kutay, U.; Schwartz, T.U. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell 2012, 149, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatau, S.B.; Hale, C.M.; Stewart-Hutchinson, P.J.; Patel, M.S.; Stewart, C.L.; Searson, P.C.; Hodzic, D.; Wirtz, D. A perinuclear actin cap regulates nuclear shape. Proc. Natl. Acad. Sci. USA 2009, 106, 19017–19022. [Google Scholar] [CrossRef] [Green Version]
- Chambliss, A.B.; Khatau, S.B.; Erdenberger, N.; Robinson, D.K.; Hodzic, D.; Longmore, G.D.; Wirtz, D. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci. Rep. 2013, 3, 1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Khatau, S.B.; Feng, Y.; Walcott, S.; Sun, S.X.; Longmore, G.D.; Wirtz, D. Actin cap associated focal adhesions and their distinct role in cellular mechanosensing. Sci. Rep. 2012, 2, 555. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; Hegele, R.A. Complex effects of laminopathy mutations on nuclear structure and function. Clin. Genet. 2019, 95, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Bruser, L.; Bogdan, S. Adherens Junctions on the Move-Membrane Trafficking of E-Cadherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a029140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, H.; Samsonov, M.; Sokabe, M. Actomyosin contractility provokes contact inhibition in E-cadherin-ligated keratinocytes. Sci. Rep. 2017, 7, 46326. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.N.; Tremblay, A.M.; Knopp, P.; White, R.B.; Urcia, R.; De Bari, C.; Zammit, P.S.; Camargo, F.D.; Wackerhage, H. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J. Cell Sci. 2012, 125 Pt 24, 6009–6019. [Google Scholar] [CrossRef] [Green Version]
- Goh, Q.; Song, T.; Petrany, M.J.; Cramer, A.A.; Sun, C.; Sadayappan, S.; Lee, S.J.; Millay, D.P. Myonuclear accretion is a determinant of exercise-induced remodeling in skeletal muscle. eLife 2019, 8, e44876. [Google Scholar] [CrossRef]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.A.; Dietz, J.M.; Jacobs, B.L.; McNally, R.M.; You, J.S.; Hornberger, T.A. Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. 2015, 589, 1491–1497. [Google Scholar] [CrossRef] [Green Version]
- Watt, K.I.; Turner, B.J.; Hagg, A.; Zhang, X.; Davey, J.R.; Qian, H.; Beyer, C.; Winbanks, C.E.; Harvey, K.F.; Gregorevic, P. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat. Commun. 2015, 6, 6048. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-Mediated Mechanotransduction in Skeletal Muscle. Front. Physiol. 2016, 7, 41. [Google Scholar] [CrossRef]
- Watt, K.I.; Judson, R.; Medlow, P.; Reid, K.; Kurth, T.B.; Burniston, J.G.; Ratkevicius, A.; De Bari, C.; Wackerhage, H. Yap is a novel regulator of C2C12 myogenesis. Biochem. Biophys. Res. Commun. 2010, 393, 619–624. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Partridge, T.A.; Matsuda, R.; Zammit, P.S. Entry of muscle satellite cells into the cell cycle requires sphingolipid signaling. J. Cell Biol. 2006, 174, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Munck, M.; Swaminathan, K.; Kapinos, L.E.; Noegel, A.A.; Neumann, S. Mutations in LMNA modulate the lamin A—Nesprin-2 interaction and cause LINC complex alterations. PLoS ONE 2013, 8, e71850. [Google Scholar] [CrossRef] [Green Version]
- Folker, E.S.; Ostlund, C.; Luxton, G.W.; Worman, H.J.; Gundersen, G.G. Lamin A variants that cause striated muscle disease are defective in anchoring transmembrane actin-associated nuclear lines for nuclear movement. Proc. Natl. Acad. Sci. USA 2011, 108, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Brosig, M.; Ferralli, J.; Gelman, L.; Chiquet, M.; Chiquet-Ehrismann, R. Interfering with the connection between the nucleus and the cytoskeleton affects nuclear rotation, mechanotransduction and myogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 1717–1728. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Lammerding, J. Keeping the LINC: The importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochem. Soc. Trans. 2011, 39, 1729–1734. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.; Mattioli, E.; Haque, F.; Antoku, S.; Columbaro, M.; Straatman, K.R.; Worman, H.J.; Gundersen, G.G.; Lattanzi, G.; Wehnert, M.; et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014, 10, e1004605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, T.P.; Cosgrove, B.D.; Heo, S.J.; Shurden, Z.E.; Mauck, R.L. Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells. Biophys. J. 2015, 108, 2783–2793. [Google Scholar] [CrossRef] [Green Version]
- Kuch, C.; Winnekendonk, D.; Butz, S.; Unvericht, U.; Kemler, R.; Starzinski-Powitz, A. M-cadherin-mediated cell adhesion and complex formation with the catenins in myogenic mouse cells. Exp. Cell Res. 1997, 232, 331–338. [Google Scholar] [CrossRef]
- Lambert, M.; Thoumine, O.; Brevier, J.; Choquet, D.; Riveline, D.; Mege, R.M. Nucleation and growth of cadherin adhesions. Exp. Cell Res. 2007, 313, 4025–4040. [Google Scholar] [CrossRef]
- Maki, K.; Han, S.W.; Hirano, Y.; Yonemura, S.; Hakoshima, T.; Adachi, T. Mechano-adaptive sensory mechanism of alpha-catenin under tension. Sci. Rep. 2016, 6, 24878. [Google Scholar] [CrossRef] [Green Version]
- Mege, R.M.; Ishiyama, N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb. Perspect. Biol. 2017, 9, a028738. [Google Scholar] [CrossRef] [Green Version]
- Weis, W.I. Cadherin structure: A revealing zipper. Structure 1995, 3, 425–427. [Google Scholar] [CrossRef] [Green Version]
- Marthiens, V.; Kazanis, I.; Moss, L.; Long, K.; Ffrench-Constant, C. Adhesion molecules in the stem cell niche—More than just staying in shape? J. Cell Sci. 2010, 123 Pt 10, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Goel, A.J.; Rieder, M.K.; Arnold, H.H.; Radice, G.L.; Krauss, R.S. Niche Cadherins Control the Quiescence-to-Activation Transition in Muscle Stem Cells. Cell Rep. 2017, 21, 2236–2250. [Google Scholar] [CrossRef] [Green Version]
- Donalies, M.; Cramer, M.; Ringwald, M.; Starzinski-Powitz, A. Expression of M-cadherin, a member of the cadherin multigene family, correlates with differentiation of skeletal muscle cells. Proc. Natl. Acad. Sci. USA 1991, 88, 8024–8028. [Google Scholar] [CrossRef] [Green Version]
- Pouliot, Y.; Gravel, M.; Holland, P.C. Developmental regulation of M-cadherin in the terminal differentiation of skeletal myoblasts. Dev. Dyn. 1994, 200, 305–312. [Google Scholar] [CrossRef]
- Zeschnigk, M.; Kozian, D.; Kuch, C.; Schmoll, M.; Starzinski-Powitz, A. Involvement of M-cadherin in terminal differentiation of skeletal muscle cells. J. Cell Sci. 1995, 108 Pt 9, 2973–2981. [Google Scholar]
- Charrasse, S.; Causeret, M.; Comunale, F.; Bonet-Kerrache, A.; Gauthier-Rouviere, C. Rho GTPases and cadherin-based cell adhesion in skeletal muscle development. J. Muscle Res. Cell Motil. 2003, 24, 309–313. [Google Scholar] [CrossRef]
- Deschenes, M. Adaptations of the neuromuscular junction to exercise training. Curr. Opin. Physiol. 2019, 10, 10–16. [Google Scholar] [CrossRef]
- Zhao, K.; Shen, C.; Lu, Y.; Huang, Z.; Li, L.; Rand, C.D.; Pan, J.; Sun, X.D.; Tan, Z.; Wang, H.; et al. Muscle Yap Is a Regulator of Neuromuscular Junction Formation and Regeneration. J. Neurosci. 2017, 37, 3465–3477. [Google Scholar] [CrossRef] [Green Version]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [Green Version]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335ra358. [Google Scholar] [CrossRef] [Green Version]
- Aure, K.; Mamchaoui, K.; Frachon, P.; Butler-Browne, G.S.; Lombes, A.; Mouly, V. Impact on oxidative phosphorylation of immortalization with the telomerase gene. Neuromuscul. Disord. 2007, 17, 368–375. [Google Scholar] [CrossRef]
- Chaouch, S.; Mouly, V.; Goyenvalle, A.; Vulin, A.; Mamchaoui, K.; Negroni, E.; Di Santo, J.; Butler-Browne, G.; Torrente, Y.; Garcia, L.; et al. Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: Validation of an exon-skipping approach to restore dystrophin in Duchenne muscular dystrophy cells. Hum. Gene Ther. 2009, 20, 784–790. [Google Scholar]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacene, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Vignier, N.; Mougenot, N.; Bonne, G.; Muchir, A. Effect of genetic background on the cardiac phenotype in a mouse model of Emery-Dreifuss muscular dystrophy. Biochem. Biophys. Rep. 2019, 19, 100664. [Google Scholar] [CrossRef]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardi, M.; Munoz-Canoves, P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Ferry, A.; Parlakian, A.; Joanne, P.; Fraysse, B.; Mgrditchian, T.; Roy, P.; Furling, D.; Butler-Browne, G.; Agbulut, O. Mechanical Overloading Increases Maximal Force and Reduces Fragility in Hind Limb Skeletal Muscle from Mdx Mouse. Am. J. Pathol. 2015, 185, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Murach, K.A.; Vechetti, I.J., Jr.; Fry, C.S.; Vickery, C.; Peterson, C.A.; McCarthy, J.J.; Campbell, K.S. MyoVision: Software for automated high-content analysis of skeletal muscle immunohistochemistry. J. Appl. Physiol. (1985) 2018, 124, 40–51. [Google Scholar] [CrossRef]
- Messeant, J.; Dobbertin, A.; Girard, E.; Delers, P.; Manuel, M.; Mangione, F.; Schmitt, A.; Le Denmat, D.; Molgo, J.; Zytnicki, D.; et al. MuSK frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance. J. Neurosci. 2015, 35, 4926–4941. [Google Scholar] [CrossRef] [Green Version]
- R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 22 October 2020).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human ribosomal protein lateral stalk subunit P0. | h-RPLPO | fw | CTCCAAGCAGATGCAGCAGA |
| rev | ATAGCCTTGCGCATCATGGT | ||
| rev | AAA-CCT-GAG-GCT-TCC-TCG-TC | ||
| siRNA1 against M-cadherin | siRNA1-cdh15 | fw | CCC-UUG-AUG-ACA-UCA-AUG-A55 |
| rev | UCA-UUG-AUG-UCA-UCA-AGG-G55 | ||
| siRNA2 against M-cadherin | siRNA2-cdh15 | fw | CAU-CGC-CGA-CUU-CAU-CAA-U55 |
| rev | AUU-GAU-GAA-GUC-GGC-GAU-G55 | ||
| siRNA3 against M-cadherin | siRNA3-cdh15 | fw | GUG-AAC-CUC-AUC-UUU-GUA-U55 |
| rev | AUA-CAA-AGA-UGA-GGU-UCA-C55 |
| Gender | LMNA Mutation | Muscle Biopsy/Age (Years) | |
|---|---|---|---|
| P1 | M | R249W | Deltoid/1 year |
| P2 | F | L380S | Deltoid/3 years |
| P3 | M | H222P | Biceps/59 years |
| C1 | M | Unknown/4 days | |
| C2 | M | Unknown/3 years | |
| C3 | M | Quadriceps/33 years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owens, D.J.; Messéant, J.; Moog, S.; Viggars, M.; Ferry, A.; Mamchaoui, K.; Lacène, E.; Roméro, N.; Brull, A.; Bonne, G.; et al. Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth. Int. J. Mol. Sci. 2021, 22, 306. https://doi.org/10.3390/ijms22010306
Owens DJ, Messéant J, Moog S, Viggars M, Ferry A, Mamchaoui K, Lacène E, Roméro N, Brull A, Bonne G, et al. Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth. International Journal of Molecular Sciences. 2021; 22(1):306. https://doi.org/10.3390/ijms22010306
Chicago/Turabian StyleOwens, Daniel J., Julien Messéant, Sophie Moog, Mark Viggars, Arnaud Ferry, Kamel Mamchaoui, Emmanuelle Lacène, Norma Roméro, Astrid Brull, Gisèle Bonne, and et al. 2021. "Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth" International Journal of Molecular Sciences 22, no. 1: 306. https://doi.org/10.3390/ijms22010306
APA StyleOwens, D. J., Messéant, J., Moog, S., Viggars, M., Ferry, A., Mamchaoui, K., Lacène, E., Roméro, N., Brull, A., Bonne, G., Butler-Browne, G., & Coirault, C. (2021). Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth. International Journal of Molecular Sciences, 22(1), 306. https://doi.org/10.3390/ijms22010306