
Cross-Predicting Essential Genes between Two Model Eukaryotic Species Using Machine Learning


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. A Selection of Strong Predictive Features of Essential Genes for CE and DM Identified by Predictions within Species
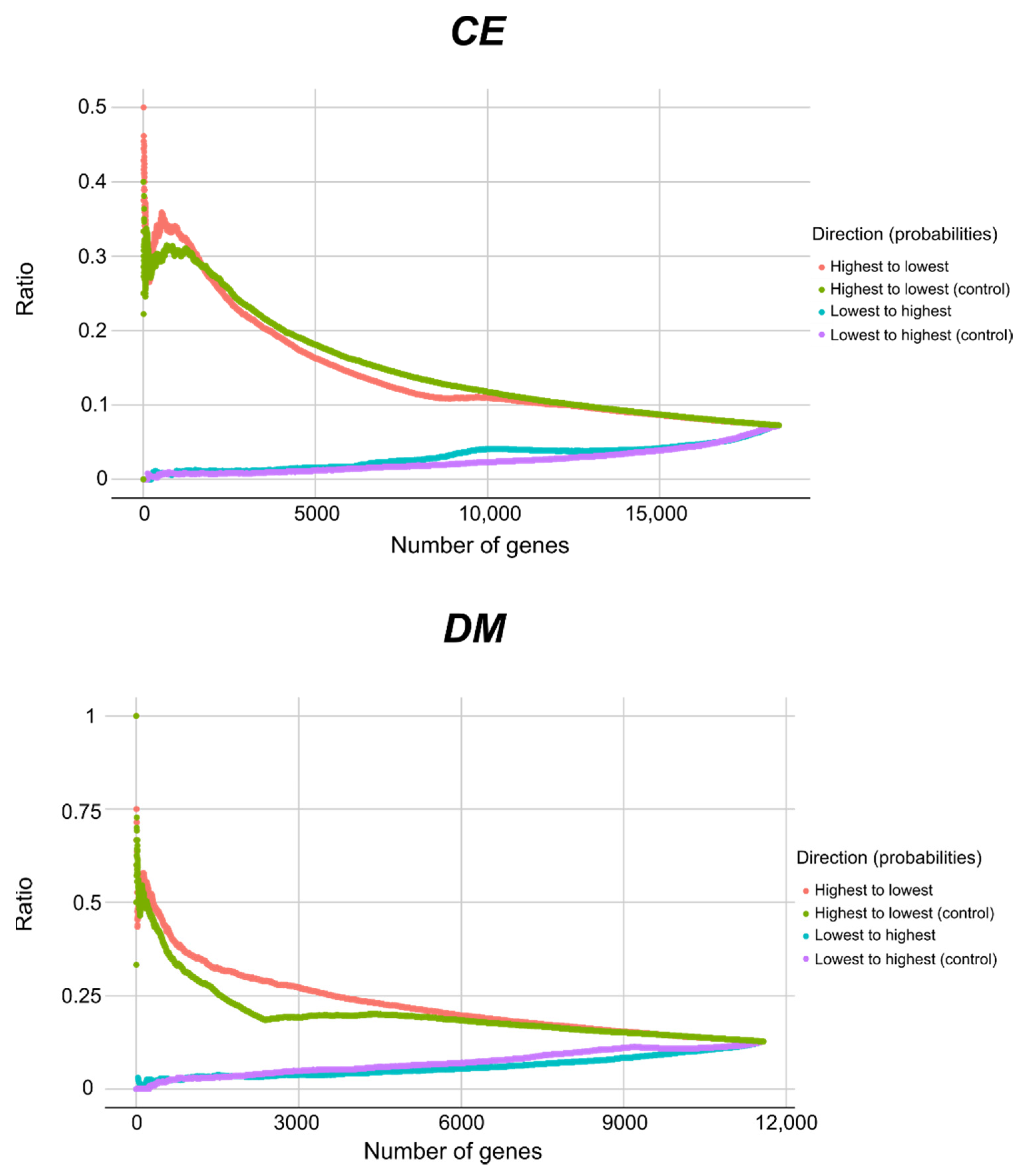
2.2. Select Features and ML Models Enable Essential Gene Predictions between CE and DM
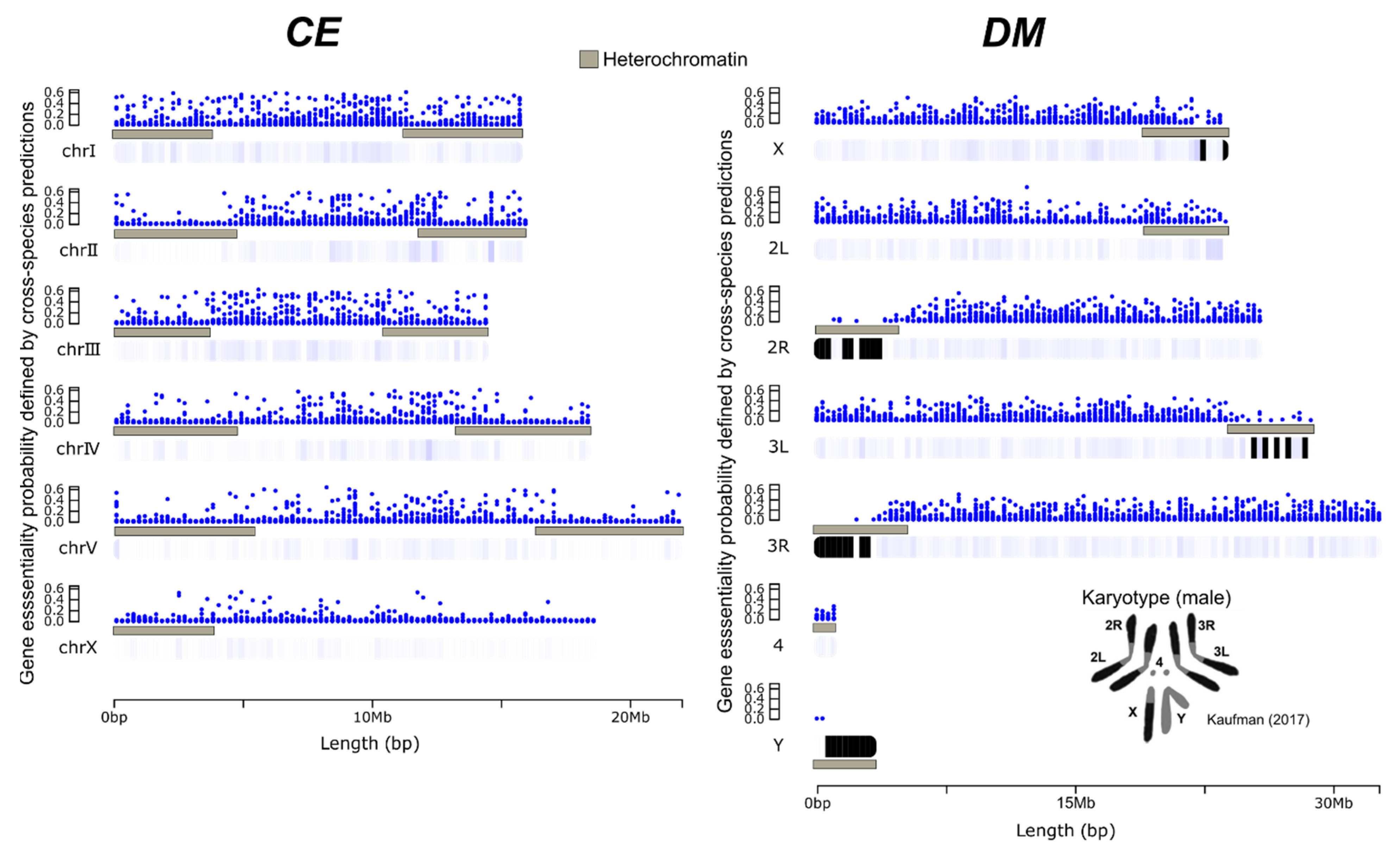
2.3. Visual Representation of Cross-Species Gene Essentiality Probabilities along the CE and DM Genomes
2.4. Gene Ontology (GO) Enrichment and Functional Clustering Analyses Confirm Important Roles of Essential Genes
3. Discussion
4. Materials and Methods
4.1. Defining Feature Sets
4.2. Feature Selection, ML Training, and Evaluation within Species Using Standardised Data
4.3. Employing and Evaluating the ML Approach for Predictions between Species
4.4. Gene Ontology Analyses with Functional Annotation Clustering
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Telford, M.J.; Bourlat, S.J.; Economou, A.; Papillon, D.; Rota-Stabelli, O. The evolution of the Ecdysozoa. Philos. Trans. R. Soc. B. Biol. Sci. 2008, 363, 1529–1537. [Google Scholar] [CrossRef]
- Abraham, E.G.; Cha, S.-J.; Jacobs-Lorena, M. Towards the genetic control of insect vectors: An overview. Entomol. Res. 2007, 37, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgerson, P.R. One world health: Socioeconomic burden and parasitic disease control priorities. Vet. Parasitol. 2013, 195, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, P.E.; Waldock, J.; Christophides, G.K.; Hemming, D.; Agusto, F.; Evans, K.J.; Fefferman, N.; Gaff, H.; Gumel, A.; LaDeau, S.; et al. Climate, environmental and socio-economic change: Weighing up the balance in vector-borne disease transmission. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370, 20130551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedges, S.B. The origin and evolution of model organisms. Nat. Rev. Genet. 2002, 3, 838–849. [Google Scholar] [CrossRef]
- Zhan, T.; Boutros, M. Towards a compendium of essential genes—From model organisms to synthetic lethality in cancer cells. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Itaya, M. An estimation of minimal genome size required for life. FEBS Lett. 1995, 362, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V. How many genes can make a cell: The minimal-gene-set concept. Annu. Rev. Genom. Hum. Genet. 2000, 1, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; Eberl, L.; Glass, J.I. Essence of life: Essential genes of minimal genomes. Trends Cell Biol. 2011, 21, 562–568. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Zhan, S.; Huang, Y. Genome editing in insects: Current status and challenges. Nat. Sci. Rev. 2019, 6, 399–401. [Google Scholar] [CrossRef]
- Zhang, X.; Acencio, M.L.; Lemke, N. Predicting essential genes and proteins based on machine learning and network topological features: A comprehensive review. Front. Physiol. 2016, 7, 75. [Google Scholar] [PubMed] [Green Version]
- Howe, K.L.; Bolt, B.J.; Cain, S.; Chan, J.; Chen, W.J.; Davis, P.; Done, J.; Down, T.; Gao, S.; Grove, C.; et al. WormBase 2016: Expanding to enable helminth genomic research. Nucleic Acids Res. 2016, 44, D774–D780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos, G.; Schroeder, A.J.; Goodman, J.L.; Strelets, V.B.; Crosby, M.A.; Thurmond, J.; Emmert, D.B.; Gelbart, W.M. FlyBase: Introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 2015, 43, D690–D697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Jin, Y.-T.; Hua, H.-L.; Wen, Q.-F.; Luo, S.; Zheng, W.-X.; Guo, F.-B. Comprehensive review of the identification of essential genes using computational methods: Focusing on feature implementation and assessment. Brief. Bioinform. 2018, 21, 171–181. [Google Scholar] [CrossRef]
- Gustafson, A.M.; Snitkin, E.S.; Parker, S.C.; DeLisi, C.; Kasif, S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genom. 2006, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Acencio, M.L.; Lemke, N. Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinforma. 2009, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xiao, W.; Hu, X. Predicting essential proteins by integrating orthology, gene expressions, and PPI networks. PLoS ONE 2018, 13, e0195410. [Google Scholar] [CrossRef] [Green Version]
- Campos, T.L.; Korhonen, P.K.; Gasser, R.B.; Young, N.D. An evaluation of machine learning approaches for the prediction of essential genes in eukaryotes using protein sequence-derived features. Comput. Struct. Biotechnol. J. 2019, 17, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, O.; Beder, T.; Oswald, M.; Oyelade, J.; Adebiyi, E.; Koenig, R. Essential gene prediction in Drosophila melanogaster using machine learning approaches based on sequence and functional features. Comput. Struct. Biotechnol. J. 2020, 18, 612–621. [Google Scholar] [CrossRef]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Predicting gene essentiality in Caenorhabditis elegans by feature engineering and machine-learning. Comput. Struct. Biotechnol. J. 2020, 18, 1093–1102. [Google Scholar] [CrossRef]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Combined use of feature engineering and machine learning to predict essential genes in Drosophila melanogaster. NAR Genom. Bioinform. 2020, 2, lqaa051. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gupta, S.; Mohmad, A.; Fular, A.; Parthasarathi, B.C.; Chaubey, A.K. Molecular tools-advances, opportunities and prospects for the control of parasites of veterinary importance. Int. J. Trop. Insect Sci. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hutter, H.; Suh, J. GExplore 1.4: An expanded web interface for queries on Caenorhabditis elegans protein and gene function. Worm 2016, 19, e1234659. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.E.; Pelz, O.; Buhlmann, S.; Kerr, G.; Horn, T.; Boutros, M. GenomeRNAi: A database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 2013, 41, D1021–D1026. [Google Scholar] [CrossRef] [Green Version]
- Garrigues, J.M.; Sidoli, S.; Garcia, B.A.; Strome, S. Defining heterochromatin in C. elegans through genome-wide analysis of the heterochromatin protein 1 homolog HPL-2. Genome Res. 2015, 25, 76–88. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, T.C. A short history and description of Drosophila melanogaster classical genetics: Chromosome aberrations, forward genetic screens, and the nature of mutations. Genetics 2017, 206, 665–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 18, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Karaiskos, N.; Wahle, P.; Alles, J.; Boltengagen, A.; Ayoub, S.; Kipar, C.; Kocks, C.; Rajewsky, N.; Zinzen, R.P. The Drosophila embryo at single-cell transcriptome resolution. Science 2017, 358, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of histone H3K4 methylation patterns. Mol. Cell 2017, 16, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Altenhoff, A.M.; Glover, N.M.; Train, C.M.; Kaleb, K.; Warwick Vesztrocy, A.; Dylus, D. The OMA orthology database in 2018: Retrieving evolutionary relationships among all domains of life through richer web and programmatic interfaces. Nucleic Acids Res. 2018, 46, D477–D485. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabási, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [Green Version]
- Doyle, M.A.; Gasser, R.B.; Woodcroft, B.J.; Hall, R.S.; Ralph, S.A. Drug target prediction and prioritization: Using orthology to predict essentiality in parasite genomes. BMC Genom. 2010, 11, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389403. [Google Scholar] [CrossRef]
- Saito, T.; Rehmsmeier, M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS ONE 2015, 10, e0118432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagus, R.; Lusa, L. Joint use of over- and under-sampling techniques and cross-validation for the development and assessment of prediction models. BMC Bioinform. 2015, 16, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative analysis of metazoan chromatin organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef] [Green Version]
- Talbert, P.B.; Henikoff, S. What makes a centromere? Exp. Cell Res. 2020, 15, 111895. [Google Scholar] [CrossRef] [PubMed]
- Cutter, A.D. Reproductive evolution: Symptom of a selfing syndrome. Curr. Biol. 2006, 18, R1056–R1058. [Google Scholar] [CrossRef] [Green Version]
- Woods, S.; Coghlan, A.; Rivers, D.; Warnecke, T.; Jeffries, S.J.; Kwon, T.; Rogers, A.; Hurst, L.D.; Ahringer, J. Duplication and retention biases of essential and non-essential genes revealed by systematic knockdown analyses. PLoS Genet. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.Y.; Zhang, J. Mouse duplicate genes are as essential as singletons. Trends Genet. 2007, 23, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Liao, G.; Brokstein, P.; Hong, L.; Carninci, P.; Shiraki, T.; Hayashizaki, Y.; Champe, M.; Pacleb, J.; Wan, K.; et al. The Drosophila gene collection: Identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 2002, 12, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washington, N.L.; Stinson, E.O.; Perry, M.D.; Ruzanov, P.; Contrino, S.; Smith, R.; Zha, Z.; Lyne, R.; Carr, A.; Lloyd, P.; et al. The modENCODE Data Coordination Center: Lessons in harvesting comprehensive experimental details. Database 2011, 2011, bar023. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Shirley Liu, X. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, T.L.; Korhonen, P.K.; Young, N.D. Cross-Predicting Essential Genes between Two Model Eukaryotic Species Using Machine Learning. Int. J. Mol. Sci. 2021, 22, 5056. https://doi.org/10.3390/ijms22105056
Campos TL, Korhonen PK, Young ND. Cross-Predicting Essential Genes between Two Model Eukaryotic Species Using Machine Learning. International Journal of Molecular Sciences. 2021; 22(10):5056. https://doi.org/10.3390/ijms22105056
Chicago/Turabian StyleCampos, Tulio L., Pasi K. Korhonen, and Neil D. Young. 2021. "Cross-Predicting Essential Genes between Two Model Eukaryotic Species Using Machine Learning" International Journal of Molecular Sciences 22, no. 10: 5056. https://doi.org/10.3390/ijms22105056
APA StyleCampos, T. L., Korhonen, P. K., & Young, N. D. (2021). Cross-Predicting Essential Genes between Two Model Eukaryotic Species Using Machine Learning. International Journal of Molecular Sciences, 22(10), 5056. https://doi.org/10.3390/ijms22105056