
Factors Associated with Mutations: Their Matching Rates to Cardiovascular and Neurological Diseases


 , ,
, ,
Abstract
:1. Introduction
2. Results
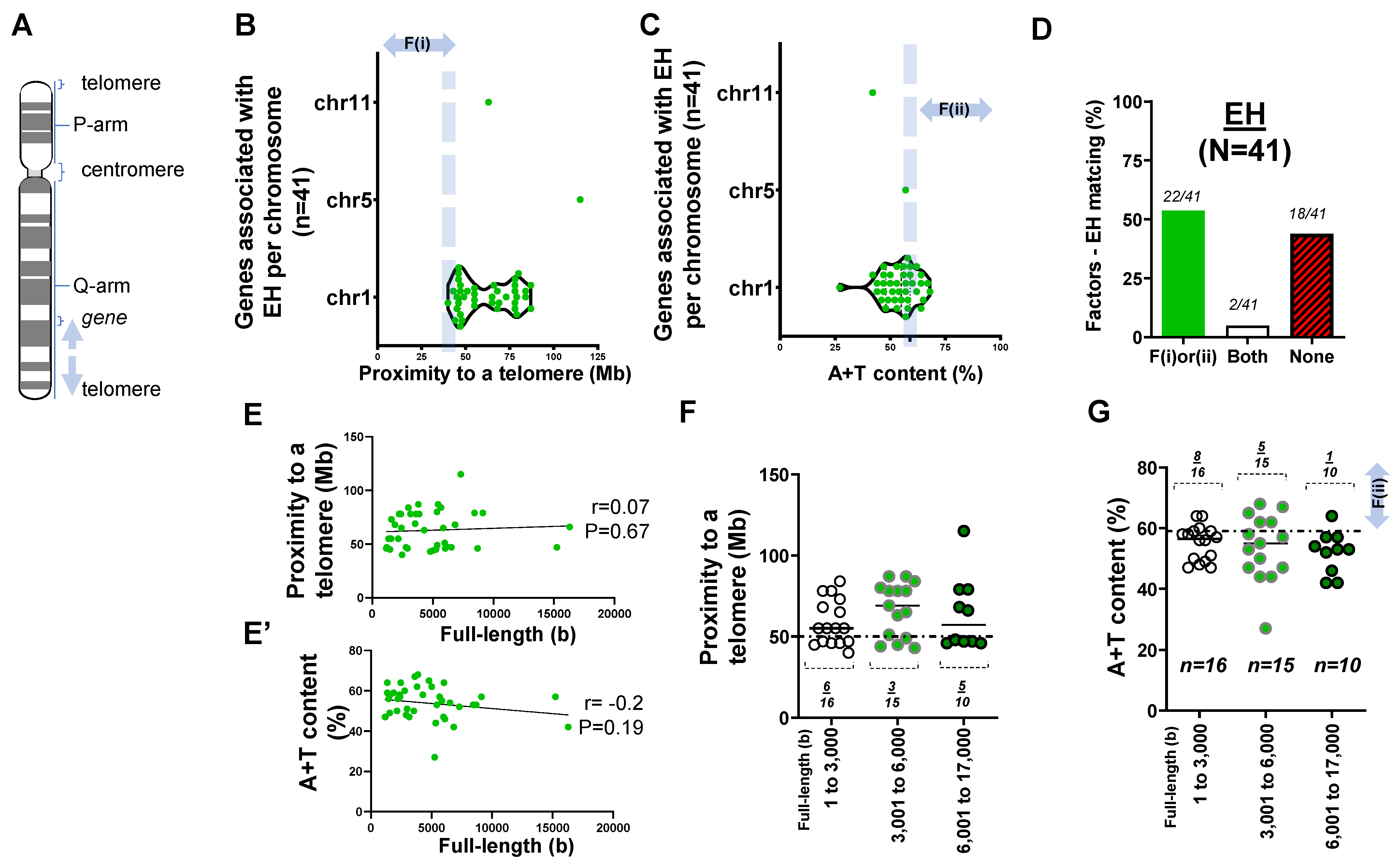
2.1. Forty-One Genes Associated with Idiopathic or Essential Hypertension (EH)
2.2. Rare Mutations and SNPs Contributing to the Genetic Architecture of Hypertension
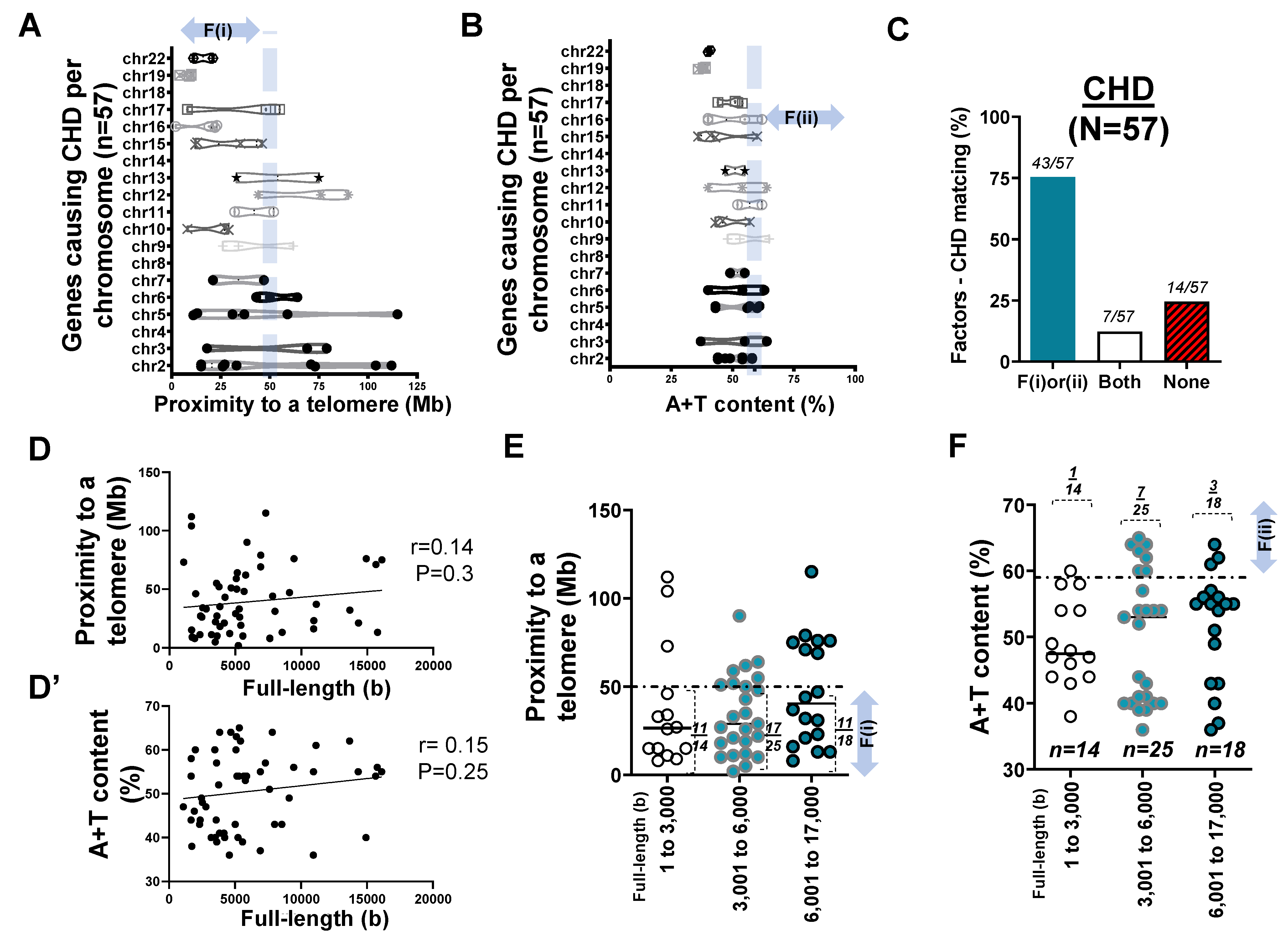
2.3. Mapping of 57 Genes Causing CHD in Human Chromosomes
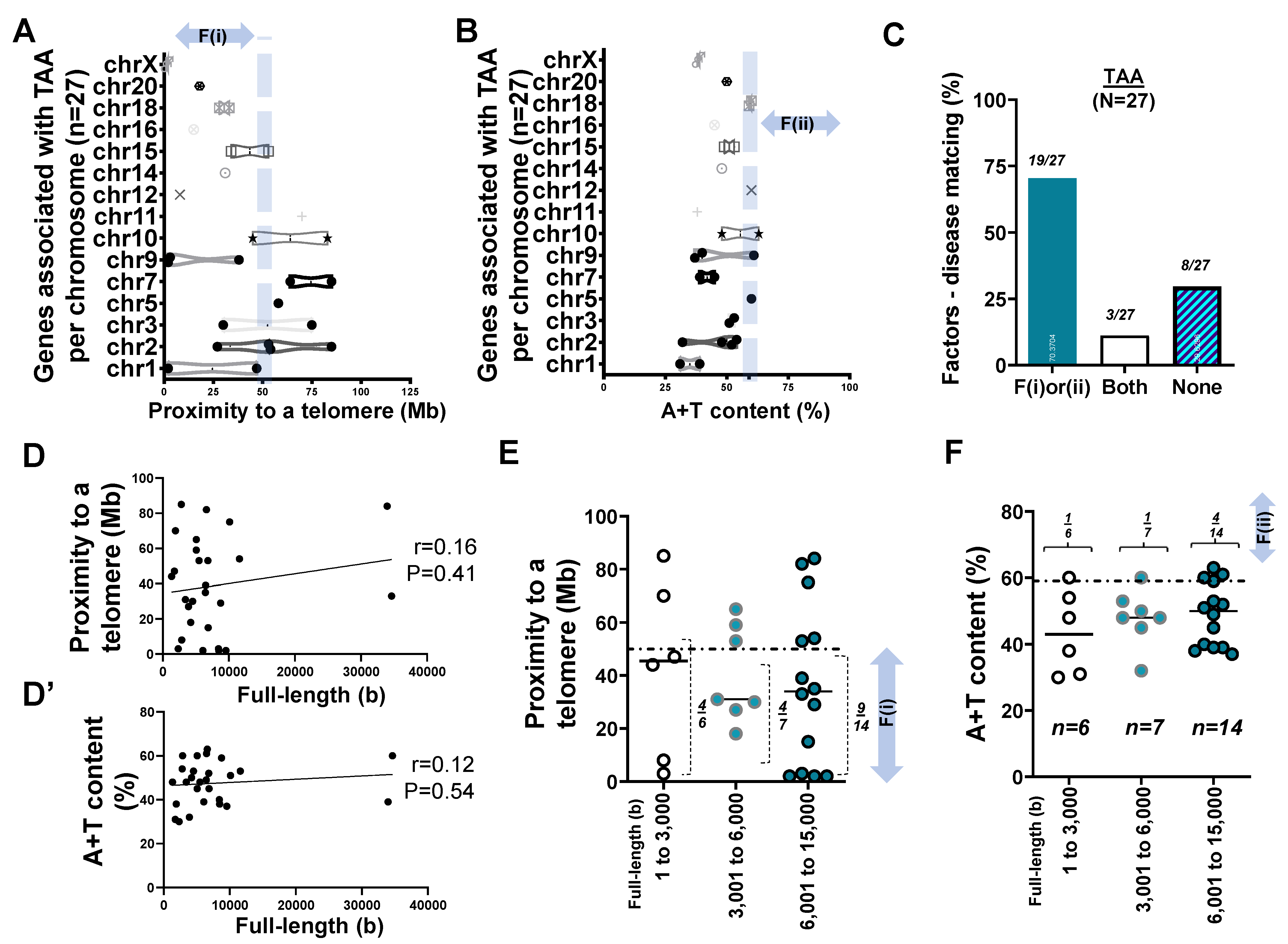
2.4. Mapping of 27 Genes Associated with TAA
3. Discussion
4. Materials and Methods
4.1. Database
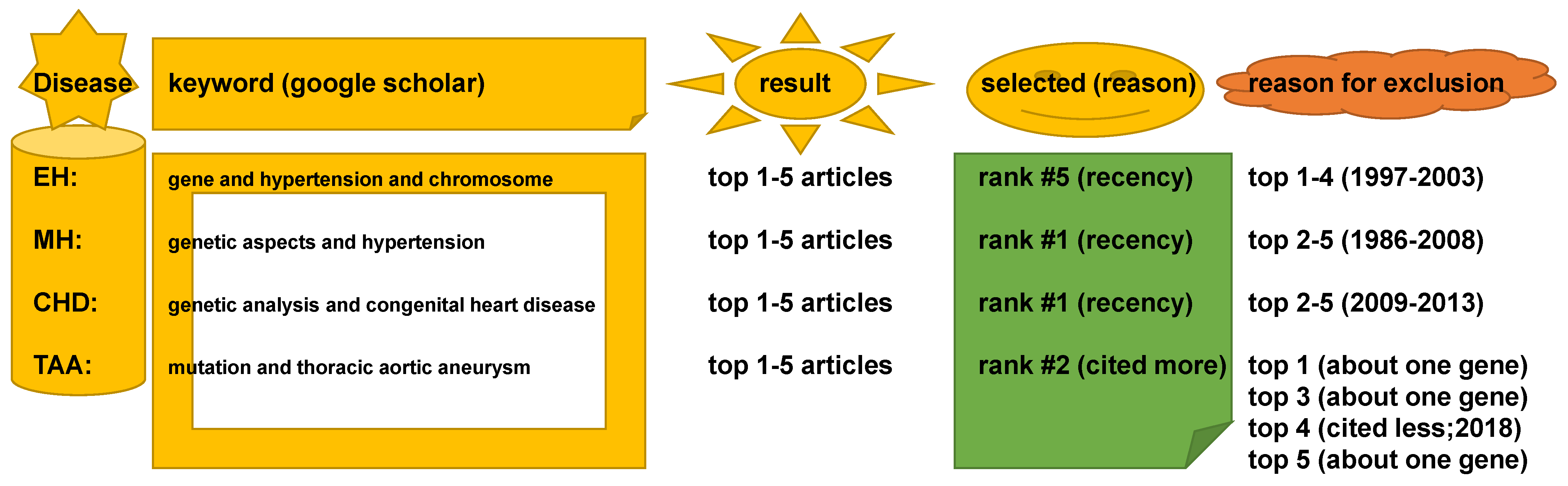
4.2. Keywords in Literature Search
4.3. Open-Source Software
- (1)
- If recombination frequency [42] is less than (≤) 50 cM, genes are linked;
- (2)
- if recombination frequency is higher than 50 cM, genes are not linked,
4.4. Data Plot and Statistical Methods
5. Conclusions
- Two factors reasonably explain the genetic mutations of cardiovascular disorders such as MH (>25 rare mutations) and the genetic architecture of hypertension (53 SNPs) at an 81% matching rate.
- Our result using the first factor—particularly in MH and the genetic architecture of hypertension—contributed by genetic variants suggests that susceptible genes located in chromosomes 18 to 22 more likely meet proximity to telomeres (<50 Mb) due to their short chromosomal lengths.
- Factor–nucleotide size relationships suggest that the full-length size of a gene associated with MH and the genetic architecture of hypertension longer than 6000 bp likely show high A+T content at >59%.
- If genes susceptible to CVD with mutations do not satisfy two criteria or the factors according to the previous suggestion [12], then neutral or deleterious mutations may appear in a region that is unlikely to be mutated, or the methods by which susceptible genes are filtered out (e.g., microsatellite markers vs. next generation sequencing) should be verified, or the contribution of environmental factors (e.g., high salt diet, body mass index, and sedentary lifestyle) might be more significant than that of genetic factors.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beaney, T.; Schutte, A.E.; Stergiou, G.S.; Borghi, C.; Burger, D.; Charchar, F.; Cro, S.; Diaz, A.; Damasceno, A.; Espeche, W.; et al. May Measurement Month 2019: The Global Blood Pressure Screening Campaign of the International Society of Hypertension. Hypertension 2020, 76, 333–341. [Google Scholar] [CrossRef]
- Butler, M.G. Pediatric hypertension: Genetics of hypertension • current status. J. Med. Liban. 2010, 58, 175–178. [Google Scholar]
- Levanovich, P.E.; Diaczok, A.; Rossi, N.F. Clinical and Molecular Perspectives of Monogenic Hypertension. Curr. Hypertens. Rev. 2020, 16, 91–107. [Google Scholar] [CrossRef]
- McKnight, I.; Hart, C.; Park, I.H.; Shim, J.W. Genes causing congenital hydrocephalus: Their chromosomal characteristics of telomere proximity and DNA compositions. Exp. Neurol. 2020, 335, 113523. [Google Scholar] [CrossRef]
- Roach, J.C.; Glusman, G.; Smit, A.F.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, H.; Sulem, P.; Kehr, B.; Kristmundsdottir, S.; Zink, F.; Hjartarson, E.; Hardarson, M.T.; Hjorleifsson, K.E.; Eggertsson, H.P.; Gudjonsson, S.A.; et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 2017, 549, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.; Huttley, G. Quantifying Influences on Intragenomic Mutation Rate. G3 (Bethesda) 2020, 10, 2641–2652. [Google Scholar] [CrossRef]
- Hellmann, I.; Prufer, K.; Ji, H.; Zody, M.C.; Paabo, S.; Ptak, S.E. Why do human diversity levels vary at a megabase scale? Genome Res. 2005, 15, 1222–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.; Hillier, L.; Eichler, E.; Zody, M.; Jaffe, D.; Yang, S.P.; Enard, W.; Hellmann, I.; Lindblad-Toh, K.; Altheide, T.; et al. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar]
- Ellegren, H.; Smith, N.G.; Webster, M.T. Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev. 2003, 13, 562–568. [Google Scholar] [CrossRef]
- Lercher, M.J.; Hurst, L.D. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002, 18, 337–340. [Google Scholar] [CrossRef]
- Nusbaum, C.; Mikkelsen, T.S.; Zody, M.C.; Asakawa, S.; Taudien, S.; Garber, M.; Kodira, C.D.; Schueler, M.G.; Shimizu, A.; Whittaker, C.A.; et al. DNA sequence and analysis of human chromosome 8. Nature 2006, 439, 331–335. [Google Scholar] [CrossRef]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Carretero, O.A.; Oparil, S. Essential hypertension. Part I: Definition and etiology. Circulation 2000, 101, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Cecelja, M.; Chowienczyk, P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc. Dis. 2012, 1, 1–10. [Google Scholar] [CrossRef]
- Yambe, M.; Tomiyama, H.; Hirayama, Y.; Gulniza, Z.; Takata, Y.; Koji, Y.; Motobe, K.; Yamashina, A. Arterial stiffening as a possible risk factor for both atherosclerosis and diastolic heart failure. Hypertens. Res. 2004, 27, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Lattanzi, S.; Brigo, F.; Silvestrini, M. Hypertension and arterial stiffness. J. Clin. Hypertens. (Greenwich) 2019, 21, 1481–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, S.; Boutouyrie, P. Arterial Stiffness and Hypertension in the Elderly. Front. Cardiovasc. Med. 2020, 7, 544302. [Google Scholar] [CrossRef]
- Hacker, A.L.; Reiner, B.; Oberhoffer, R.; Hager, A.; Ewert, P.; Muller, J. Increased arterial stiffness in children with congenital heart disease. Eur. J. Prev. Cardiol. 2018, 25, 103–109. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Tellides, G. Central artery stiffness and thoracic aortopathy. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H169–H182. [Google Scholar] [CrossRef]
- Boczar, K.E.; Boodhwani, M.; Beauchesne, L.; Dennie, C.; Chan, K.L.; Wells, G.A.; Coutinho, T. Aortic Stiffness, Central Blood Pressure, and Pulsatile Arterial Load Predict Future Thoracic Aortic Aneurysm Expansion. Hypertension 2021, 77, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Lear, S.A.; Hu, W.; Rangarajan, S.; Gasevic, D.; Leong, D.; Iqbal, R.; Casanova, A.; Swaminathan, S.; Anjana, R.M.; Kumar, R.; et al. The effect of physical activity on mortality and cardiovascular disease in 130,000 people from 17 high-income, middle-income, and low-income countries: The PURE study. Lancet 2017, 390, 2643–2654. [Google Scholar] [CrossRef]
- Lusis, A.J. Genetics of atherosclerosis. Trends Genet. 2012, 28, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Lusis, A.J.; Mar, R.; Pajukanta, P. Genetics of atherosclerosis. Annu. Rev. Genom. Hum. Genet. 2004, 5, 189–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, L.D.; Tang, N.L.S.; Xu, Z.F.; Xu, J. Genes Regulate Blood Pressure, but “Environments” Cause Hypertension. Front. Genet. 2020, 11, 580443. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Silva, G.V.; Drager, L.F. What Is the Most Common Cause of Secondary Hypertension? An Interdisciplinary Discussion. Curr. Hypertens. Rep. 2020, 22, 101. [Google Scholar] [CrossRef]
- Zentner, D.; James, P.; Bannon, P.; Jeremy, R. Familial Aortopathies—State of the Art Review. Heart Lung Circ. 2020, 29, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Avila, H.A.; Vallejo, M.; Martinez-Garcia, M.; Hernandez-Lemus, E. Comorbidity Networks in Cardiovascular Diseases. Front. Physiol. 2020, 11, 1009. [Google Scholar] [CrossRef]
- Firouzi, M.; Sherkatolabbasieh, H.; Nezami, A.; Shafizadeh, S. Congenital Heart Disease in non-Diabetic Large-for-Gestational-Age (LGA) Neonates. Cardiovasc. Hematol. Disord. Drug Targets 2020. [Google Scholar] [CrossRef]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the length, weight and GC content of the human genome. BMC Res. Notes 2019, 12, 106. [Google Scholar] [CrossRef]
- Misteli, T. The long reach of telomeres. Genes Dev. 2014, 28, 2445–2446. [Google Scholar] [CrossRef] [Green Version]
- McKnight, T.D.; Shippen, D.E. Plant telomere biology. Plant. Cell 2004, 16, 794–803. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Caulfield, M.; Dominiczak, A.F. Genetic and molecular aspects of hypertension. Circ. Res. 2015, 116, 937–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennstedt, C.; Chaoui, R.; Korner, H.; Dietel, M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: Results of a seven year necropsy study. Heart 1999, 82, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownstein, A.J.; Kostiuk, V.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2018 Update and Clinical Implications. Aorta (Stamford) 2018, 6, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Brownstein, A.J.; Ziganshin, B.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: An Update and Clinical Implications. Aorta (Stamford) 2017, 5, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinholo, T.F.; Brownstein, A.J.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2019 Update and Clinical Implications. Aorta (Stamford) 2019, 7, 99–107. [Google Scholar]
- Chang, Y.P.; Liu, X.; Kim, J.D.; Ikeda, M.A.; Layton, M.R.; Weder, A.B.; Cooper, R.S.; Kardia, S.L.; Rao, D.C.; Hunt, S.C.; et al. Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am. J. Hum. Genet. 2007, 80, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Ritter, E.; Gebhardt, C.; Salamini, F. Estimation of recombination frequencies and construction of RFLP linkage maps in plants from crosses between heterozygous parents. Genetics 1990, 125, 645–654. [Google Scholar] [CrossRef]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef]
- Hackett, J.A.; Feldser, D.M.; Greider, C.W. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001, 106, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Zschenker, O.; Reynolds, G.; Miller, D.; Murnane, J.P. Effect of telomere proximity on telomere position effect, chromosome healing, and sensitivity to DNA double-strand breaks in a human tumor cell line. Mol. Cell. Biol. 2010, 30, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastbacka, J.; de la Chapelle, A.; Kaitila, I.; Sistonen, P.; Weaver, A.; Lander, E. Linkage disequilibrium mapping in isolated founder populations: Diastrophic dysplasia in Finland. Nat. Genet. 1992, 2, 204–211. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiovascular System | MH [35] | CHD [37] | TAA [39] | EH [41] |
| Factor–disease match | 81% | 75% | 70% | 53% |
| No. of genes (loci) FL–A+T correlation Short–Mid–Long (−3000)(−6000)(−17,000 bp) Relative type of gene size | n = 79 sig. 22:29:28 (1:1.3:1.3) longer | n = 57 NS 14:25:18 (1:1.8:1.3) longer | n = 27 NS 6:7:14 (1:1.2:2.3) longer | n = 41 NS 16:15:10 (1.6:1.5:1) shorter |
| CNS | CH [4] | AD [4] | fPD [4] | |
| Factor–disease match | >90% | 84% | 59% | |
| No. of genes (loci) FL–A+T correlation Short–Mid–Long (−3000)(−6000)(−15,000 bp) Relative type of gene size | n = 108 sig. 40:39:29 (1.4:1.3:1) shorter | n = 70 sig. 39:22:9 (4.3:2.4:1) shorter | n = 17 sig. 7:5:5 (1.4:1:1) shorter | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, H.B.; McKnight, I.; Raines, R.; Hijazi, A.; Hart, C.; Lee, C.; Kim, D.-G.; Li, W.; Lee, P.H.U.; Shim, J.W. Factors Associated with Mutations: Their Matching Rates to Cardiovascular and Neurological Diseases. Int. J. Mol. Sci. 2021, 22, 5057. https://doi.org/10.3390/ijms22105057
Lucas HB, McKnight I, Raines R, Hijazi A, Hart C, Lee C, Kim D-G, Li W, Lee PHU, Shim JW. Factors Associated with Mutations: Their Matching Rates to Cardiovascular and Neurological Diseases. International Journal of Molecular Sciences. 2021; 22(10):5057. https://doi.org/10.3390/ijms22105057
Chicago/Turabian StyleLucas, Hannah B., Ian McKnight, Regan Raines, Abdullah Hijazi, Christoph Hart, Chan Lee, Do-Gyoon Kim, Wei Li, Peter H. U. Lee, and Joon W. Shim. 2021. "Factors Associated with Mutations: Their Matching Rates to Cardiovascular and Neurological Diseases" International Journal of Molecular Sciences 22, no. 10: 5057. https://doi.org/10.3390/ijms22105057
APA StyleLucas, H. B., McKnight, I., Raines, R., Hijazi, A., Hart, C., Lee, C., Kim, D. -G., Li, W., Lee, P. H. U., & Shim, J. W. (2021). Factors Associated with Mutations: Their Matching Rates to Cardiovascular and Neurological Diseases. International Journal of Molecular Sciences, 22(10), 5057. https://doi.org/10.3390/ijms22105057