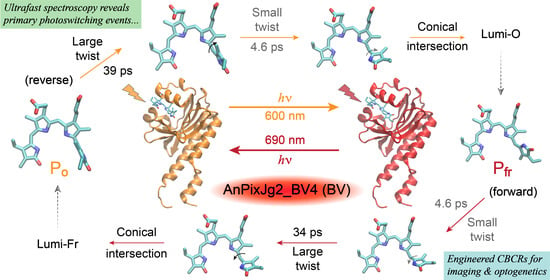
2.2. Time-Resolved Electronic Spectroscopy Tracks Reversible Photoswitching of PCB and BV Cofactors in the AnPixJg2_BV4 Pocket
The time-resolved electronic spectra of CBCRs in buffer solution using ultrafast laser pulses (
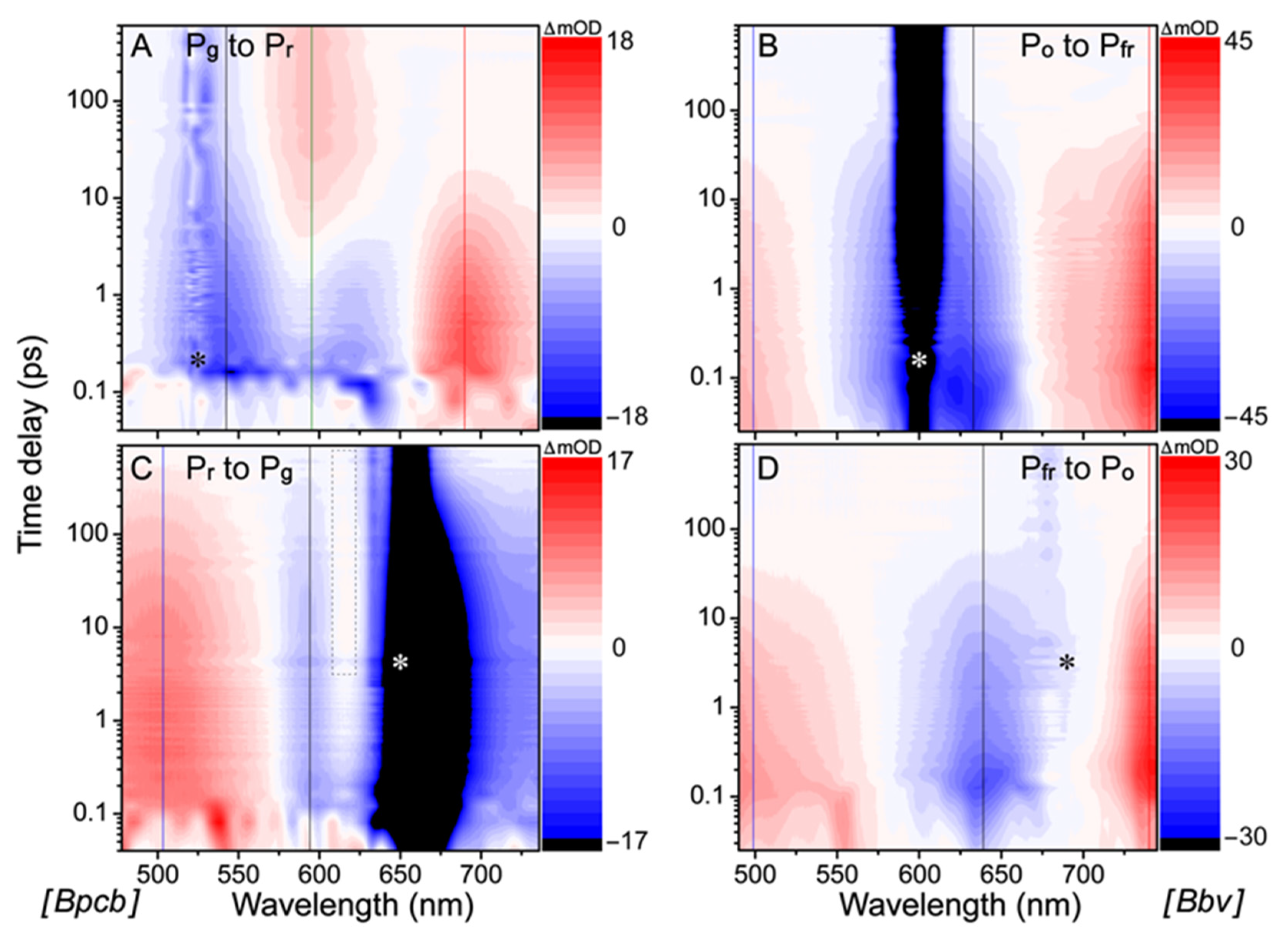
Figure 2) track the cofactor molecular population as it evolves on the excited-state potential energy surface (PES). There are four characteristic features in the fs-TA spectra: excited-state absorption (ESA), hot ground-state absorption (HGSA), ground-state bleaching (GSB), and stimulated emission (SE). Both ESA and HGSA bands are positive whereas GSB and SE bands are negative. The GSB feature overlaps with the ground state absorption band (see
Figure 1D–F) which distinguishes it from the typically red-shifted SE and HGSA features. Notably, the measured TA signal intensity for Bpcb photoswitching processes (ca. ±18 mOD, OD is optical density,
Figure 2A,C) is similar to that for Apcb at a similar sample concentration (OD ≈ 0.8 per mm at the P
r absorption peak) [
15], whereas the less concentrated Bbv (OD ≈ 0.4 per mm at the P
fr absorption peak; see
Table 1 in
Section 2.1 above) displays much stronger TA intensity (~2–2.5 fold,
Figure 2B,D). This result implies a larger excited-state transition oscillator strength [
21,
22] of the more conjugated BV cofactor than PCB cofactor in the CBCR pocket (see
Figure 1) [
18].
For corroboration, the raw experimental time-stacked fs-TA spectra (
Supplementary Figure S1) exhibit the ultrafast dynamics of key electronic features, and the probe-dependent signal intensity (±5 nm of peak wavelength, see
Supplementary Figure S2) least-squares fits yield multiple exponentials with characteristic time constants and amplitude weights [
23]. Since the protein cofactor photoswitching process typically involves excited state intermediates undergoing parallel and/or sequential transitions, the observed broad spectral features could be highly overlapped so the retrieved time constants from the probe-dependent fits may be convoluted or combined into an averaged value. Therefore, it is imperative to analyze and compare the least-squares fits with the associated time constants and amplitude weights for multiple TA marker bands to identify common features for the excited-state chromophores (see each panel in
Supplementary Figure S2 for a specific photoswitching process in various CBCR samples under study).
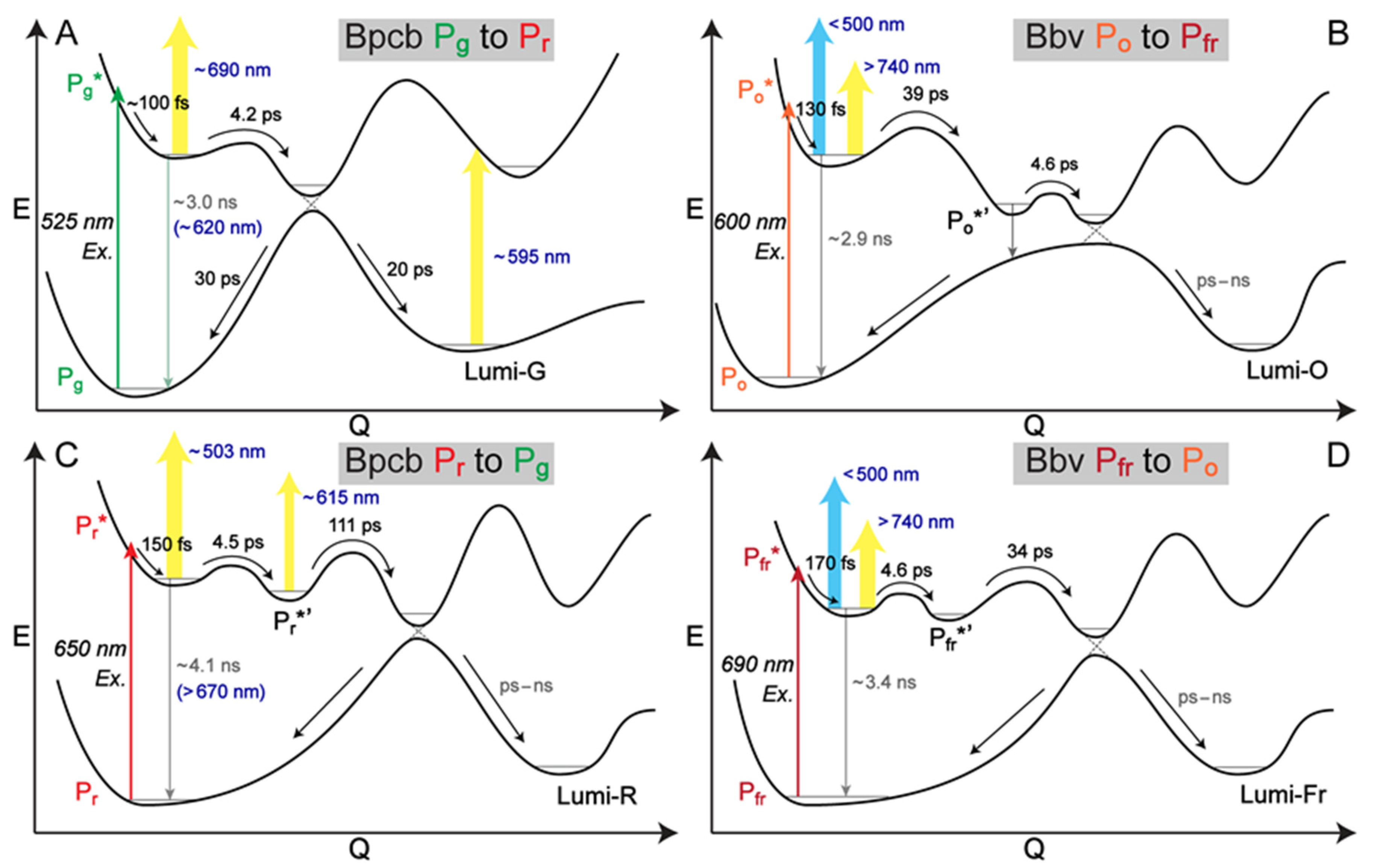
To help deconvolute the underlying cofactor species with pertinent time constants, global analysis was implemented to analyze TA data within the entire detection spectral and time window, allowing a holistic representation of the excited state electronic dynamics. The global analysis method typically employs two different models, evolution-associated difference spectrum (EADS) and decay-associated difference spectrum (DADS) [
24]. In short, EADS uses multiple sequential states to model the spectra whereas DADS uses a parallel model with each species decaying with distinct lifetimes, which could provide complementary information for the complex TA data with overlapping spectral bands. The −2 ps (time delay, meaning that the probe pulse precedes the actinic pump pulse by 2 ps, hence serving as a useful background) TA spectrum was subtracted from the subsequent data traces to remove the strong scattering signal (some residuals can still be seen in
Figure 2). During global analysis in Glotaran [
25], the pump scattering was either set to zero if the feature was in the middle of the window or the window was trimmed when the scattering was at the edge so it would not interfere with the least-squares fits of the chromophore spectral features (
Figure 3). Notably, the retrieved components from global analysis do not necessarily correspond to distinct molecular species, but rather, they represent a mathematical description of the spectral data with characteristic separable components and time constants in capturing the chromophore electronic dynamics.
The fs-TA spectral 2D-contour plot (
Figure 2A) and global analysis results (
Figure 3A,E) of Bpcb P
g → P
r photoconversion exhibit three major features that appear at the photoexcitation time zero: ~542 nm GSB (matching the steady-state absorption peak of P
g state in
Figure 1E), 620 nm SE, and 690 nm ESA. These features correspond to the P
g* (the asterisk denotes excited state) population. There is also rise of a positive feature around 595 nm that persists beyond the typical excited-state lifetime, hence it was assigned as an HGSA band of Lumi-G [
15]. The 542 nm GSB band shows recovery dynamics of ~350 fs (10%), 3.5 ps (45%), 31 ps (29%), and 1 µs (16%) (
Supplementary Figure S2A, black trace). The representative 1 µs component was used to model the long-lived population that does not return to original ground state within our detection time window. The most pronounced 690 nm ESA band shows clear decay dynamics with ~400 fs (9%), 4 ps (68%), and 27 ps (23%) time constants (
Supplementary Figure S2A, red trace), which is accompanied by a clear rise of the 595 nm HGSA that has been attributed to the Lumi-G species of PCB cofactor in the “hot” electronic ground state [
15]. Due to spectral overlap in the visible spectral region, the probe-dependent fit of the 595 nm band yields a ~200 fs (25%) and 3.4 ps (37%) decay of the initial negative feature (
Supplementary Figure S2A, green trace, reminiscent of the adjacent GSB band dynamics) and a 21 ps (11%) rise of the Lumi-G HGS species that becomes apparent after ~4 ps (see the summary PES scheme below). The ~1 µs (27%) decay component was necessary for the fit since the long-lived signal eventually diminishes well beyond our current detection time window (see Methods in
Section 3.3 below; a good separation of time constants allows the retrieval of longer time constants) [
15].
Global analysis of the TA spectra for the photoinduced Bpcb P
g → P
r conversion requires five components to achieve a satisfactory fit, wherein the initial component fits a large coherent artifact with <5 fs lifetime (much lower than our cross-correlation time of ~120 fs [
15,
26]) so it was removed from the figure to focus on major electronic dynamics of the protein cofactor (see
Figure 3A,E). The retrieved lifetimes were <100 fs, 4.2 ps, 20 ps, and 3 ns that largely match the probe-dependent fits (
Supplementary Figure S2A). The sub-ps time constant is known to be Franck-Condon (FC) relaxation as the photoexcited wavepacket moves across the PES after electronic excitation [
15,
19,
27,
28]. This ultrafast step can be seen as clear and distinct features emerge in EADS (
Figure 3A) and DADS (
Figure 3E, black to blue trace). The 4.2 ps component likely tracks a partial twisting motion of the D ring in P
g* as it moves toward a CI, consistent with literature and the well-accepted D-ring twist along the photoswitching coordinate [
15,
29]. This interpretation is also supported by the loss of P
g* ESA and SE bands at ~690 and 620 nm (
Figure 3A,E, blue to green trace), and the associated large amplitude weight that suggests a dominant relaxation pathway to the ground state. After the CI passage, the population diverges into two pathways. First, the twisted P
g* can continue to twist and reach the Lumi-G state as the primary photoproduct species, which is evident by the ~21 ps rise of the Lumi-G HGSA band (
Figure 3A,E, green to magenta trace;
Supplementary Figure S2A, green trace). Second, the twisted P
g* can reverse-twist back to P
g in the ground state as indicated by the ~31 ps GSB band recovery (
Supplementary Figure S2A, black trace). The ~3 ns component from global analysis and ~1 µs component from the probe-dependent fits are representative of a weak fluorescence process and further protein-pocket-facilitated cofactor motions as Lumi-G fully converts to P
r in the electronic ground state [
15,
30].
The Bpcb P
r → P
g TA spectra (
Figure 2C) and global analysis (
Figure 3B,F) show clear P
r* modes with an ESA band around ~520 nm and two GSB bands at 594 and 650 nm, with the latter two bands matching the main ground state absorption peak and its bluer vibronic shoulder (
Figure 1E, red trace). The strong scattering from 650 nm actinic pump made the probe-dependent fits in that region unreliable (see black shade in
Figure 2 and orange asterisk in
Supplementary Figure S1C). We thus fit the cleaner 594 nm GSB band recovery dynamics with ~150 fs (34%), 4.7 ps (20%), 118 ps (30%), and 1 ns (16%) time constants (
Supplementary Figure S2C, black trace). Meanwhile, the 503 nm ESA band exhibits similar intensity decay time constants of ~4.8 ps (13%), 254 ps (57%), and 2.5 ns (6%), but also requires a 28 ps (24%) component for a satisfactory fit (
Supplementary Figure S2C, blue trace). The retrieval of two intermediate time constants (28 and 254 ps) instead of one 118 ps time constant (from GSB recovery) or 111 ps (from global analysis of the spectra within the detection window, see below) could be due to more complex dynamics when higher-lying electronic excited states are involved (e.g., the 503 nm ESA band due to the S
1 → S
n transition) [
20,
31], although the general time scale of ~110–120 ps is consistent for the PCB cofactor excited-state relaxation along the S
1 PES.
Notably, there is a discernible rise and decay of a positive band around ~615 nm (see
Figure 2C, dashed rectangle box) which resembles the fs-TA data of Apcb [
15] and could be attributed to a slightly twisted excited-state intermediate (P
r*’). To reduce the pump scattering effect and confirm the delayed onset of this specific ESA band, we performed a control experiment using a dilute Bpcb sample (OD ≈ 0.2/mm at the P
r state absorption peak, about a quarter of the Bpcb sample concentration listed in
Table 1 and presented in
Figure 2C) and observed clear dynamics of a ~618 nm band with a much reduced scattering signal to the red side (
Supplementary Figure S3). The 743 nm SE band shows similar decay dynamics to the bluer GSB band at 594 nm (
Supplementary Figure S2C). The overall cofactor excited-state decay dynamics of Bpcb are faster than Apcb (see
Section 2.5 below for a detailed comparison), suggesting that smaller residues in the chromophore vicinity of AnPixJg2_BV4 mutant [
18] allow for faster ring-twisting events (likely accompanied by local environment relaxation) of the PCB cofactor [
32].
Global analysis of the Bpcb P
r → P
g TA spectra yields ~150 fs, 4.5 ps, 111 ps, and 4.1 ns lifetimes. As discussed above, the 150 fs component reflects FC dynamics of the P
r* species while the 4.5 ps component tracks the P
r* → P
r*’ transition. This key assignment was made previously in the Apcb P
r → P
g conversion by a pronounced rise and decay of the 616 nm ESA band of P
r*’ species [
15]. However, the more significant overlap with a stronger GSB band (versus that in Apcb) makes it more difficult to reliably fit the weak 615 nm P
r*’ ESA band dynamics of Bpcb. Nevertheless, transient spectral signatures of the P
r*’ state is evident from global analysis (
Figure 3B,F, blue to green trace, see the black arrow) and the pertinent ~620 nm ESA band decays away on the 111 ps time scale, which likely corresponds to the non-equilibrium molecular movement toward a peaked CI [
27,
33] that can repopulate the original ground state and convert to the Lumi-R state [
15,
29]. Notably, the CI is also evident from the dominant loss of excited-state features with the 111 ps lifetime (
Figure 3B,F, green to magenta trace), while the longer-lived 4.1 ns component with an SE band redder than ~670 nm (
Figure 2C, with a more pronounced SE peak at ~743 nm in the dilute CBCR sample in
Supplementary Figure S3) is likely owing to the fluorescence transition from P
r* [
15,
29]. Overall, the P
r → P
g conversion is less efficient than the P
g → P
r conversion since the majority of excited-state species return to the original ground state [
15,
29,
34], in accord with the observed longer lifetimes and the lack of prominent new TA features emerging at late times after 650 nm photoexcitation (see
Figure 2C and
Figure 3B).
For comparison, the fs-TA spectra of the newly engineered Bbv exhibit similar features for both the P
o → P
fr and P
fr → P
o transitions (
Figure 2B,D), which differ from the PCB cases (
Figure 2A,C). Although the observed TA bands are shifted in wavelength, they both exhibit a blue ESA band at the edge of our spectral window (<500 nm), a GSB band in the middle region (around 633 and 639 nm for P
o → P
fr and P
fr → P
o transitions, respectively, see
Supplementary Figure S1B,D), and a strong red ESA band (>740 nm). An expanded spectral window to the bluer or redder probe regions is required to determine the exact locations for the blue and red ESA peaks; however, the increased similarity between the TA signatures of BV’s P
fr and P
o states versus PCB’s P
r and P
g states indicates the major effect of the intrinsic cofactor electronic structure, which likely causes the largely unchanged TA bands that are consistent with much less energy separation between the two conformers in BV (624 and 699 nm in
Figure 1F, ~1719 cm
−1) versus PCB (542 and 650 nm in
Figure 1E, ~3066 cm
−1). These common TA features in
Figure 2B,D mainly track the P
o* and P
fr* electronic dynamics because they appear at the photoexcitation time zero and decay away with no clear wavelength shifts. It is notable that the P
fr GSB band around 690 nm overlaps with the pump scattering that is largely removed for TA analysis (
Supplementary Figure S1D). Interestingly, all the least-squares fits of excited state dynamics show similar time constants for both conversions of the BV cofactor (
Supplementary Figure S2B,D), which differs significantly from the PCB cofactor counterparts (
Supplementary Figure S2A,C). For the P
o → P
fr conversion, the probe-dependent fits expose ~125 fs (43%), 4.6 ps (18%), and 32 ps (39%) time constants for the 499 nm ESA band, ~150 fs (48%), 4.6 ps (17%), 39 ps (34%), and 2.9 ns (1%) time constants for the 633 nm GSB band, and ~125 fs (27%), 4.6 ps (23%), 39 ps (49%), and 2.9 ns (1%) time constants for the 740 nm ESA band (see
Supplementary Figure S2B). Similar dynamic components are apparent for these TA bands that report on the common excited-state decay pathways of a presumably homogeneous P
o* population [
35,
36,
37] as discussed in detail below.
For the P
fr → P
o conversion, the corresponding probe-dependent fits expose ~125 fs (35%), 4.8 ps (27%), 39 ps (37%), and 3.4 ns (1%) time constants for the 499 nm ESA band, ~129 fs (50%), 4.6 ps (21%), 37 ps (28%), and 3.4 ns (1%) for the 639 nm GSB band, and ~130 fs (31%), 4.9 ps (31%), 36 ps (34%), and 3 ns (4%) time constants for the 740 nm ESA band (see
Supplementary Figure S2D), which reflect the common excited-state decay pathways of a presumably homogeneous P
fr* population. Note that such a systematic analysis of independent measurements on reversible photoswitching processes of the same CBCR sample (e.g., Bbv with specific light irradiation conditions listed in
Table 1 in
Section 2.1 above) substantiates the robustness of the aforementioned time constants and amplitude weights for the underlying dynamic components retrieved after an actinic pump. As further corroboration, global analysis of the P
o → P
fr (
Figure 3C,G) and P
fr → P
o (
Figure 3D,H) transitions also output similar time constants of ~130 and 170 fs, 4.6 and 4.6 ps, 39 and 34 ps, and 2.9 and 3.4 ns, respectively. Similar to the Bpcb P
g → P
r conversion case (
Figure 3A,E), the fit for a large coherent artifact was removed in the reversible photoswitching between P
o and P
fr states of Bbv to retrieve the intrinsic underlying time constants along the excited-state PES of the protein cofactor. Moreover, though the EADS show the blue trace (4.6 ps lifetime) decaying into green trace (~35 ps lifetime,
Figure 3C,D), the corresponding DADS show the green trace intensity higher than blue trace in both ESA regions (below 550 nm and above 720 nm,
Figure 3G,H). This finding is indicative of some deviations from a simple kinetic model that the 4.6 ps component precedes the 35 ps component (see below).
To confirm these key observations and results for the Bbv photoswitching mechanisms, we performed the following control experiments: (1) Fs-TA data sets were collected at least twice with the freshly prepared protein samples on different days in the laser lab to ensure reproducibility. (2) The steady-state absorption spectra of the P
o and P
fr conformers were taken before and after every TA experiment to confirm that the protein samples maintain their photoswitching capabilities. (3) Control TA experiments using 600 nm actinic pump with 600 nm LEDs (
Supplementary Figure S4A) and 704 nm actinic pump with the 650 nm longpass-filtered tungsten lamp (
Supplementary Figure S4B) were conducted to ensure that the TA data in
Figure 2B,D were not just collecting the same conformer or mixed species. (4) A visual confirmation was achieved that the actinic pump could convert the protein samples by holding the sample cuvette in the beam path before it was focused [
15]. (5) A redder actinic pump at 704 nm was used and similar TA features were observed for the P
fr → P
o transition (see
Supplementary Figure S5 and
Figure 2D). With all these strategic, useful, and interwoven control experiments substantiating the aforementioned results, the observed similar dynamics of the P
o → P
fr and P
fr → P
o transitions infer a very alike or even “purely” reversible excited-state pathway of the BV cofactor in AnPixJg2_BV4.
The major differences between the aforementioned reversible photoswitching dynamics are the amplitude weights of the two intermediate components retrieved on the ps time scale. The P
o → P
fr conversion (
Supplementary Figure S2B) shows about half the weight for the ~5 ps component (18%, 17%, and 23%) compared to the ~35 ps component (39%, 34%, and 49%) for the blue ESA, GSB, and red ESA bands, respectively. In contrast, for the corresponding TA marker bands, the P
fr → P
o conversion (
Supplementary Figure S2D) shows approximately equal weights for the ~5 ps (27%, 21%, and 31%) and ~35 ps (37%, 28%, and 34%) components. This experimental finding poses two interesting questions. (1) Why does Bbv exhibit similar photoswitching dynamics in both directions whereas the Apcb and Bpcb P
g → P
r and P
r → P
g conversions show different excited-state pathways? (2) Why are the amplitude weights of two largely conserved intermediate ps time constants the major differentiating factor between the Bbv P
o → P
fr and P
fr → P
o photoconversion?
2.3. Altered Excited-State Dynamics Arise from an Interplay between PCB or BV Cofactor and AnPixJg2 or AnPixJg2_BV4 on Ultrafast Time Scales
To answer these fundamental questions, we first examined the differences between PCB and BV cofactors in the AnPixJg2_BV4 pocket. The location-shifted BV cofactor in the BV4 pocket moves the A ring closer to its H-bonding partners Asp291 and Trp289 [
18]. The distance between the cofactor A ring nitrogen atom and the Asp291 oxygen atom decreases from ~1.95 Å in the Apcb P
r state crystal structure (PDB ID: 3W2Z) [
8] to 1.76 Å in the Bbv P
fr state crystal structure (PDB ID: 5ZOH at 1.60 Å resolution) [
18]. The distance between the cofactor A ring carbonyl oxygen and the Trp289 nitrogen is ~0.21 Å closer in Bbv (2.76 Å) than Apcb (2.97 Å). The closer H-bonding partners and the extra electron density in the A ring would effectively restrict it from large twisting motions, while a slight twist of the A ring has been reported as a component in the photoconversion process [
15,
37,
38]. By better stabilizing the A ring inside the protein pocket, more of the photoconverting energy could be localized at the D ring with a reversible clockwise/counterclockwise twist (i.e., along the same isomerization pathway but in opposite directions) by reducing motions away from the D ring.
As the D ring twists, there are two dihedral angles that correlate with the conversion between P
fr and P
o conformers. The dihedral closer to the D ring (D-dihedral) likely causes a smaller twisting motion than the dihedral closer to the C ring (C-dihedral, see
Figure 1A–C), since the D-dihedral twist is a volume-conserving motion whereas the C-dihedral twist likely increases the volume [
39,
40,
41]. Upon excitation of P
fr to P
fr*, the conjugated double bonds acquire more single-bond character and vice versa [
33,
42,
43], in accord with previous studies showing that the C15=C16 bond (directly connecting to the D ring) is responsible for the major twisting between the 15
Z/
E conformers [
44,
45,
46]. By analyzing both dihedral angles for the available Apcb P
r state and Bbv P
fr state crystal structures, the C-dihedral (CCCC) around the bridge single bond (36.08° vs. 51.12°) is more twisted in Bbv whereas the D-dihedral (CCCN) around the bridge double bond (26.96° vs. 0.83°) is more twisted in Apcb from the pyrrole ring plane [
8,
15,
18]. The BV4 mutations, specifically the more critical F308T and I336V, help to accommodate the shifted cofactor around the D ring due to less bulky residues [
18]. Notably, if we attribute the observed 4.6 and 34 ps in the Bbv P
fr → P
o photoconversion (
Figure 3D) to the small-scale D-dihedral and large-scale C-dihedral twisting motions, respectively, we could then expect a smaller and larger time constant from the cofactor TA dynamics in Apcb due to the aforementioned starting geometry. Indeed for the C-dihedral twist, we observed a 217 ps component for the Apcb P
r → P
g transition [
15], and a 111 ps component for the Bpcb P
r → P
g transition (
Figure 3B) likely owing to an intermediate geometry of the D ring in between the Apcb P
r and Bbv P
fr conformers. In contrast, for the D-dihedral twist on the few ps time scale, it is less sensitive to the initial geometry alone mainly due to its close interaction with an adjacent Val336 or Ile336 (in Bbv, Bpcb or Apcb, see
Figure 1A–C) that could affect the faster time constant (in preparation for the subsequent larger twist motion) [
20,
47].
It is likely that Bpcb also takes advantage of the extra space around the cofactor and adopts a slightly shifted orientation from Bbv. The larger area around the D ring also explains the overall faster photoswitching dynamics in Bpcb than Apcb [
32]. The slightly slower ~4 ps component in Bpcb P
g → P
r than ~3 ps in Apcb P
g → P
r conversion [
15] can be explained by the shifted PCB cofactor in the AnPixJg2_BV4 pocket which would bring the D ring closer to Val336 and somewhat hinder the initial smaller twist (i.e., D-dihedral). Although the smaller twisting motion becomes slower, the larger twist (i.e., C-dihedral) occurs faster in the P
g → P
r photoconversion of Bpcb (~20 ps) than Apcb (~30 ps), both retrieved from global analysis of the corresponding TA spectra after 525 nm photoexcitation. We note that the larger twist occurs in the electronic ground state past the CI, likely owing to the metastable P
g* species that undergoes a more downhill and efficient process than the more “trapped” P
r* species that requires both twisting processes in the electronic excited state [
15,
29]. Interestingly for the latter case, the P
r → P
g photoconversion of Bpcb (~4.5 and 111 ps) also occurs faster than Apcb (~13 and 217 ps) involving presumably small and large ring-twisting motions of the protein cofactor, both retrieved from global analysis of the corresponding TA spectra after 650 nm photoexcitation [
15].
For Bbv, because the photoswitching pathways are fully reversible with largely conserved time constants, the amplitude weights from the TA signal intensity fits become crucial in gaining mechanistic insights into the excited-state pathways. Since Bpcb does not exhibit such a reversible TA pattern during photoswitching, the observed “fully reversible” pathway seems to be unique to Bbv (i.e., both AnPixJg2_BV4 pocket and BV cofactor are needed). The probe-dependent least-squares fits implement a parallel model of multiple processes with a common time zero, although it does not mean that the fastest component occurs first. By inspecting the Apcb and Bpcb photoswitching scheme, the 3–4 and 20–30 ps components likely represent the aforementioned small and large twisting processes, respectively, although the detailed assignments to excited- or ground-state dynamics need to come from the TA spectral analysis. The amplitude weight of each time constant is representative of how dominant the associated pathway is for energy dissipation. Since the Bbv TA bands emerge around photoexcitation time zero and then decay, while the peak wavelength remains largely unshifted up to 900 ps (
Figure 2B,D), the observed TA band intensity dynamics likely track the excited-state population change along the ring-twisting coordinates. Notably, a larger twist could cause a larger change of the electronic transition oscillator strength for the resultant conformation than that of a smaller twist, which may contribute to the apparent TA band intensity decay dynamics. We thus focus on the change of amplitude weight ratios instead of exact weights for ensuing analysis.
In the P
fr → P
o conversion of Bbv, the equal weights of the ~5 ps and 35 ps components (
Supplementary Figure S2D) suggest that a 5 ps smaller twisting motion would populate the P
fr*’ state before undergoing the 35 ps larger twisting motion en route to the CI. The less energy-consuming and more facile 5 ps twist could reduce the chances of other nonradiative pathways that compete with the 35 ps twist, while the initial small twist out of the more stabilized P
fr state may be required to help the D ring move away from the adjacent Val336 to allow more room for the subsequent large twist in the excited state (
Figure 1C). In the P
o → P
fr conversion of Bbv, an unexpected observation that the ~35 ps larger twist being twice as dominant as the 5 ps smaller twist implies that the larger twisting motion occurs first. The more energy-consuming and less facile 35 ps twist would bring the excited state closer to the ground state, allowing for more nonradiative processes to compete with the smaller twist. This would effectively cause the weight of the smaller twisting component to drop in comparison with the larger twist [
20,
21]. After the smaller twist, the remaining population could pass through a CI and continue to undergo cofactor ring twist and the surrounding protein pocket relaxation to reach the isomerized state (see
Section 2.5 below and Graphical Abstract for illustration). We note that if we attributed the ~5 and 35 ps to inhomogeneous species of P
o* undergoing excited-state energy relaxation in parallel, then it would be presumptuous to explain the same time constants from two inhomogeneous subpopulations of P
fr* because the two conformer states have different cofactor conformations in their respective surrounding protein pockets upon electronic excitation [
8,
38].
In both Bbv conversions, most of the population returns to the original ground state. The lack of a long-lived GSB recovery is evident (
Supplementary Figure S2B,D) that per excitation pulse, the TA signal from any photoconverted population is likely within our experimental signal-to-noise ratio. Although having an apparently lower photoconversion efficiency than Apcb, Bbv still exhibit a fast photoconversion and can operate effectively under cw or ambient light sources for potential optogenetic applications [
3,
7,
17].
2.4. Ground State Femtosecond Stimulated Raman Spectroscopy Sheds Light on Structural Factors That Rationalize the Unique Reversible Photoswitching Pathways in Bbv
To help elucidate the atomic structures of Bpcb and Bbv conformers, GS-FSRS experiments were performed to obtain the vibrational signatures of Apcb, Bpcb, and Bbv (
Figure 4). Respective LEDs were implemented to collect the Apcb P
r and P
g (
Figure 4A), Bpcb P
r and P
g (
Figure 4B), as well as Bbv P
fr and P
o conformers (
Figure 4C). With the pre-resonance ps 803 nm Raman pump, the fs broadband Raman probe on the Stokes side was used to effectively prevent notable dispersive line shapes of the cofactor chromophore [
20,
48]. The collected spectra in
Figure 4A match our previous experimental results on Apcb using various Raman pump wavelengths at 792, 678, and 596 nm with Raman probe on the Stokes or anti-Stokes sides [
15]. As a control experiment, we also collected the anti-Stokes FSRS spectra with a bluer Raman probe than the Raman pump (
Supplementary Figure S6) [
22,
48,
49], which exhibit much stronger peaks due to the resonance enhancement achieved by the Raman probe with respect to the ground state absorption peaks of P
fr (699 nm) and P
o (624 nm), further confirming the vibrational peak frequencies in S
0 [
28,
48,
50,
51]. Using the Gaussian software [
52], density functional theory (DFT) calculations at B3LYP level with 6–31G(d,p) basis sets were performed on the BV cofactor to visualize the vibrational normal modes (see Experimental Methods in
Section 3.4 below). A frequency scaling factor 0.97 was applied to match the DFT-calculated spectrum to the GS-FSRS spectrum of the Bbv P
fr conformer (
Supplementary Figure S7). The discrepancy in the Raman peak intensities is likely due to the DFT calculations only treating the cofactor quantum mechanically in vacuo without the protein pocket. More accurate hybrid quantum mechanics/molecular mechanics (QM/MM) methods would help model the CBCR protein with the embedded cofactor as a whole [
37,
45,
53,
54] which could complement our experimental capabilities in ultrafast laser spectroscopy. The description of major vibrational modes of the P
fr state can be seen in
Supplementary Table S1.
Overall, Apcb, Bpcb, and Bbv share marker bands common to bilin photosensors at ~1051 and 1466 cm
−1 [
35,
55]. PCB and BV cofactors exhibit similar vibrational motions with small frequency shifts (
Figure 4). The 1051 cm
−1 mode is localized to the D-ring deformation with strong C–H sidechain wagging. The 1466 cm
−1 is attributed to the B and C ring-deformation and CH
3 rocking. Both of these modes are stabilized by the conserved His322, Trp289, and Tyr352 in the CBCR pocket [
18]. The overall higher intensity of the red-absorbing species (P
r in Apcb and Bpcb, and P
fr in Bbv) is attributed to better resonance enhancement with the 803 nm Raman pump. Moreover, the similar peak intensity at ~1050 cm
−1 for P
o and P
fr of Bbv while most other modes display reduced intensities in P
o can be rationalized by the vibronic coupling matrix that yields the mode-dependent change of resonance conditions [
15,
20], which indicates the importance of light-induced D-ring motions that connect these conformers. Key differences between the vibrational spectra of PCB (
Figure 4A,B) and BV (
Figure 4C) can be seen by the red-shifted modes around 660 and 800 cm
−1, and in the 1200–1400 cm
−1 region.
The ~660 cm
−1 peak region reveals key differences in electronic conjugation between the PCB and BV cofactors. This Raman mode involves collective C–C and C–H wagging motions in the conjugated cofactor ring system. A frequency redshift of 10 cm
−1 from the ~669 cm
−1 mode in Apcb and Bpcb (
Figure 4A,B) to 659 cm
−1 mode in Bbv (
Figure 4C) is rationalizable by two extra double bounds in the A and D rings of Bbv cofactor (see
Figure 1C). In Bbv, the peak doublet around 825 cm
−1 shows a clear blueshift of the lower-frequency mode from P
fr (797 cm
−1) to P
o (809 cm
−1). Since the associated vibrational motions involve N–H wagging from the A, B, and C rings with C–H wagging on the CD bridge [
15,
55], the frequency blueshift is likely due to a twisted D ring that disrupts the H-bonding of Trp289 to A ring. In Apcb and Bpcb, the lack of a strong peak around 800 cm
−1 could be attributed to a different H-bonding network of the A ring with reduced electric polarizability (one less double bond). In the 1200–1400 cm
−1 region, Bbv exhibits a prominent 1225 cm
−1 mode. In Bpcb and Apcb, this mode is weak but displays a noticeable redshift from Apcb to Bpcb (~1244 to 1225 cm
−1, see the tilted arrow from
Figure 4A,B). In both PCB and BV calculations [
15], since this mode involves delocalized N–H rocking and AB bridge C–H rocking motions, the clear peak enhancement in Bbv (
Figure 4C) further corroborates the shifted, stronger H-bonded A ring in the more conjugated BV cofactor. The redshift in Bpcb compared to Apcb is also consistent with the BV4 mutations that allow the PCB cofactor to adopt a slightly shifted position inside the protein pocket and establish better H-bonding interactions, yet the largely unchanged Raman peak intensity around 1235 cm
−1 indicates that the mode-dependent electric polarizability is dictated by the intrinsic chromophore electronic structure (see the clear peak intensity ratio and line width change from
Figure 4A,B to
Figure 4C) [
20,
29].
2.5. Contrasting Excited-State Potential Energy Surfaces for Reversible Photoswitching of PCB and BV Cofactors in the AnPixJg2_BV4 Pocket
With all the correlated electronic and vibrational signatures from steady-state to time-resolved regime [
15,
20], we could sketch the overall reversible photoswitching pathways of Bpcb and Bbv (
Figure 5). Bpcb adopts the similar reaction scheme as Apcb as the BV4 mutations do not affect the major pathways of the embedded cofactor. The less bulky residues (particularly Thr308 and Val336,
Figure 1B) in Bpcb allow the D ring more room to twist, decreasing the excited-state dynamics time constants [
15,
36]. In the Bpcb P
g → P
r conversion (
Figure 5A), the P
g* species undergoes FC relaxation on the ~100 fs time scale, then approaches a CI on the ~4.2 ps time scale. The slightly shifted cofactor in Bpcb makes the initial twist slightly slower than that in Apcb (~2.7 ps) [
15]. After the CI, the cofactor population likely continues twisting to the converted Lumi-G state or twisting back to P
g on the 20–30 ps time scales. This step is evident by a ~20 ps component in the Lumi-G rise (HGSA at ~595 nm in
Figure 2A) and a ~30 ps component in the GSB recovery (
Supplementary Figure S2A). As for the opposite P
r → P
g conversion (
Figure 5C), after FC relaxation the P
r* species transitions into a twisted P
r*’ intermediate with a 4.5 ps time constant, which requires further ring twists into a CI on the 111 ps time scale. The P
r*’ state can be seen by the transient rise and decay of a weak ESA band around 615 nm (
Figure 2C and
Supplementary Figure S3), corroborated by a stronger ESA band around 616 nm with similar dynamics for the same PCB cofactor inside the AnPixJg2 pocket (i.e., Apcb) [
15].
On the long time scale, since the 3–4 ns minor component from global analysis of Bpcb (
Figure 3A,B,E,F) is an average value of all the long processes going beyond our current time window of ~1 ns, the pertinent contributors could be fluorescence from P
g* species (see the 620 nm SE band in
Figure 2A and PES in
Figure 5A) or P
r* species (see the >670 nm SE band in
Figure 2C and PES in
Figure 5C), and the long-lasting photoproduct state that does not return to the original ground state on this time scale (e.g., Lumi-G in
Figure 5A and Lumi-R in
Figure 5C). In analogy, the similar ~3 ns minor component from global analysis of Bbv (
Figure 3C,D,G,H) could involve weak fluorescence from P
o* and P
fr* species plus the long-lasting photoproduct states Lumi-O and Lumi-Fr in
Figure 5B and 5D, respectively. It is interesting to note that these minor components with 1–4% amplitude weights were consistently retrieved from both the red ESA band (~740 nm) and GSB band (633–639 nm) of Bbv (
Supplementary Figure S2B,D), indicating that a small percentage of the excited-state population goes from the excited state to the original ground state on the ~3 ns time scale (a typical radiative transition lifetime), which could be due to the aforementioned weak fluorescence component. The absence of a prominent SE band could be due to spectral overlap with the strong and broad ESA band on the red side (
Figure 2B,D and
Figure 3C,G,D,H), and any mismatch between the ESA and GSB dynamics could imply some deviations from a direct S
1-to-S
0 transition. These long-time-scale processes could benefit from future in-depth studies with other experimental and computational techniques (see below) that are beyond the scope of this work.
Notably, the engineered Bbv undergoes a more “purely” reversible photoswitching pathway as reflected by the highly conserved excited-state time constants from either conformer of the cofactor. The P
fr → P
o conversion (
Figure 5D) adopts a similar scheme to the P
r → P
g conversion of the PCB samples from the thermally equilibrated state to the metastable state. The similarly weighted ~4.6 and 34 ps time constants infer that the 4.6 ps small-scale twisting motion occurs prior to the 34 ps larger twist, which is characteristic of the D-dihedral change acting as a preparatory step [
28,
56] for the C-dihedral change (see details in
Section 2.3 above). In contrast, starting from the metastable state with a more downhill relaxation, the higher-weighted ~39 ps larger twisting motion could generate a P
o*’ species that is energetically closer to the ground state, which allows for other nonradiative processes (depicted by a vertically downward gray arrow in
Figure 5B) to more effectively compete with the 4.6 ps small-scale twist leading into to a CI region before the Lumi-O formation. Given this kinetic scheme, we do not expect a significant accumulation of transient P
o*’ species since the 4.6 ps outgoing rate is much faster than the 39 ps incoming rate, which likely explains the lack of clear TA features from the P
o*’ species. As a result, the observed TA dynamics (
Figure 2B) mainly reflect the ultrafast multi-exponential decay of P
o* species (
Figure 5B), consistent with kinetic models with reversible reactions connecting all the transient species. In particular, the DADS feature with a 4.6 ps lifetime (
Figure 3G) does not reflect the P
o*’ species per se, instead, it mathematically tracks the 4.6 ps kinetic component (along the P
o*’-to-CI pathway) with a similar spectral profile as the initial ~130 fs and subsequent 39 ps components (i.e., P
o* species) as shown in the EADS analysis (
Figure 3C).
Recent reports on the ultrafast dynamics of a red/far-red bathy bacteriophytochrome named PaBphP that binds BV inside the GAF domain (part of a classical PAS-GAF-PHY architecture) [
32,
36] provide a useful comparison. The forward
15ZP
r →
15EP
fr photoconversion utilizes a two-step process on the excited state with a 2–207 ps local environment (e.g., protein residue sidechains, internal/trapped water molecules [
55,
56,
57], elaborate H-bonding networks) relaxation and a 375 ps isomerization to the CI [
32]. For comparison, the reverse P
fr → P
r photoconversion (with higher bond strain/distortion to release) goes through an excited-state bifurcation on the much faster ~1 and 4 ps time scales where both wavepackets reach the CI [
36]. The pertinent few-ps photoisomerization step around the C15=C16 bond that significantly shifts the cofactor D-ring with respect to a nearby protein pocket residue is consistent with our assignment of the 3–5 ps component to the D-dihedral/small-scale twisting motion (see
Section 2.3 above). By evaluating the conformational differences between P
fr and P
r states of PaBphP via X-ray crystallography, the BV cofactor exhibits distinct conformations of the A, B, C, and D rings [
58], which underlie the significant difference between forward and reverse pathways mediated by different residues in the extensively rearranged H-bonding network. In sharp contrast, stabilizing the A, B, and C rings through largely conserved H-bonding and π-π stacking (e.g., between Trp289 and D ring) in the BV4 pocket [
18] could therefore allow a rather unique and “purely” reversible photoconversion pathway of the BV cofactor (between the P
o and P
fr states of Bbv,
Figure 1F) as observed in this work (
Figure 2 and
Figure 3,
Supplementary Figures S1 and S2). Notably, the focus here is the elucidation of reversible photoswitching between distinct (not necessarily pure) conformers in the protein pocket.
The more homogeneous, thermally equilibrated populations of Bpcb and Bbv could also be related to the protonation state of the adjacent His322 [
37]. QM/MM calculations indicate that the His322 protonation state is a trigger for structural heterogeneity. In the crystal structure of the P
fr state of AnPixJg2_BV4 [
18], His322 shares two H-bonding partners: one is the B ring propionate, the other is a local water molecule that also H-bonds to the D ring nitrogen site (–NH group). The His318Tyr mutation in BV4 removes the original H-bond between His318 and the B ring propionate in AnPixJg2 [
8] and would thus increase the H-bonding interaction between B ring and His322 [
18]. For the other H-bonding partner, since both PCB and BV cofactors undergo some position shift in the BV4 pocket, the D ring likely has more tendency to move away from His322. The resultant singly protonated state of His322 (as an H-bond donor to the B ring propionate) would support a more homogeneous cofactor population [
37], which is evident from the “native” steady-state electronic absorption spectra that show Bbv (
Figure 1F, black trace) being more homogeneous than Bpcb (
Figure 1E, black trace). The homogeneous population also supports Bbv in adopting a sequential kinetic model instead of a parallel model to undergo excited-state electronic and structural relaxation of the BV cofactor with continuous active-site motions in S
1 [
30,
32], followed by slower thermal relaxation events of the chromophore and protein matrix in the electronic ground state (S
0). By exciting a mostly homogeneous ground state population, it is unlikely to have the transient wavepacket branch into different pathways with notably different time constants (e.g., ~5 and 35 ps) that both reach the CI.
Meanwhile, the P
o conformer of Bbv is likely more homogeneous due to its higher absorption peak than the P
g conformers of Apcb and Bpcb (
Figure 1F orange trace vs.
Figure 1D,E green traces, all relative to their corresponding redder-absorbing peaks), because a larger extinction coefficient could be indicative of a more stabilized P
o state of Bbv. For corroboration, implementing the BV4 mutation to AnPixJg4 (the fourth GAF domain of AnPixJ) leads to a rapid P
o-to-P
fr dark reversion that is ~400-fold faster than that of AnPixJg2_BV4 (Bbv), while the relative P
o/P
fr absorption peak intensity ratio (0.65, peak wavelengths at 628 nm/700 nm) [
18] is indeed smaller in AnPixJg4_BV4 than that in Bbv (0.85, peak wavelengths at 624 nm/699 nm,
Figure 1F). The BV-binding efficiency of AnPixJg4_BV4 (44%) is also significantly lower than AnPixJg2_BV4 (75%). Further investigation of AnPixJg4_BV4 using a similar combination of fs-TA and GS-FSRS to this work could help to unveil the mechanistic origin for the fourth GAF domain to exhibit greatly accelerated dark reversion on the ground state, shedding light on the effect of cofactor-protein-pocket interplay on the apparent TA features versus the thermally equilibrated P
fr state. Knowledge about the effects of specific attributes around the cofactor and how they contribute to the overall photoswitching dynamics would enable the protein engineers to effectively tailor these photoswitching proteins for specific applications.
To further decipher the multidimensional [
20,
56,
57] photoswitching dynamics, we expect the improved molecular dynamics (MD) simulation methods with higher accuracy and longer duration [
26,
37,
55,
57,
59] and other spectroscopic techniques to paint a holistic and accurate portrait as the molecular system is excited, photoconverting, and relaxing back to its thermally stable dark-adapted state [
29,
55]. Since the metastable light-adapted P
o and P
g crystal structures are challenging to obtain, QM/MM methodology has advanced in closely matching the calculated absorption spectra of protein conformers with their experimental counterparts, including recent successes on a similar red/green absorbing CBCR named Slr1393g3 with its P
r and P
g crystal structures both solved [
38,
54]. By implementing similar QM/MM calculations on Bbv, more accurate vibrational and structural insights into the P
o conformer could be gained to augment our current understanding of the P
fr conformer (
Figure 4 and
Table S1) with its available crystal structure [
18]. Moreover, a systematic calculation of the excited-state free energies of the BV cofactor with various D-ring twisting geometries and intermediates (see
Section 2.3 above) in the protein pocket can shed more light on the ultrafast reversible photoswitching pathway between the P
o and P
fr conformers of AnPixJg2_BV4.
In addition, an expanded spectroscopic toolset from ultrafast regime (typically fs-to-ns time scales as reported here) to the more macroscopic time scales (ns-to-ms or longer) could better identify long-lived or later intermediates (e.g., Lumi-G and Lumi-R in Apcb and Bpcb, and Lumi-O and Lumi-Fr in Bbv), which may dictate the forward/reverse reaction speed and yield [
5,
29,
60,
61]. These Lumi states are likely similar to the photoproduct species in our current experiments on the ns time scale (
Figure 5), however, they require further protein relaxation and chromophore pocket rearrangement on the electronic ground state to eventually reach the final product state. For example, a TA setup with pulsed (100 ns) LED-based broadband probe has expanded the time window from ns to ms and identified two intermediates between Lumi-G
2 and P
r after 532 nm excitation of P
g of NpR6012g4 [
62], and recent applications in conjunction with fs-TA were able to elucidate a series of meta states that emerge after the Lumi state formation in AnPixJg2 as well as NpR6012g4 with a strong sequence homology (both are canonical red/green CBCRs) [
61]. In aggregate, by combining multiple techniques such as TA, FSRS, QM/MM, X-ray crystallography, and NMR, the resultant comprehensive information can delineate the complex photoconversion of CBCRs and other photosensing proteins as they convert from one state to another on various time scales. Such a more complete understanding of the working mechanisms of these fascinating nanomachines can help more bioengineers to better predict, design, prepare, and tune the next generation of bioimaging and optogenetic toolsets to advance life sciences and improve human health.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}