
Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D

 , , and
, , and 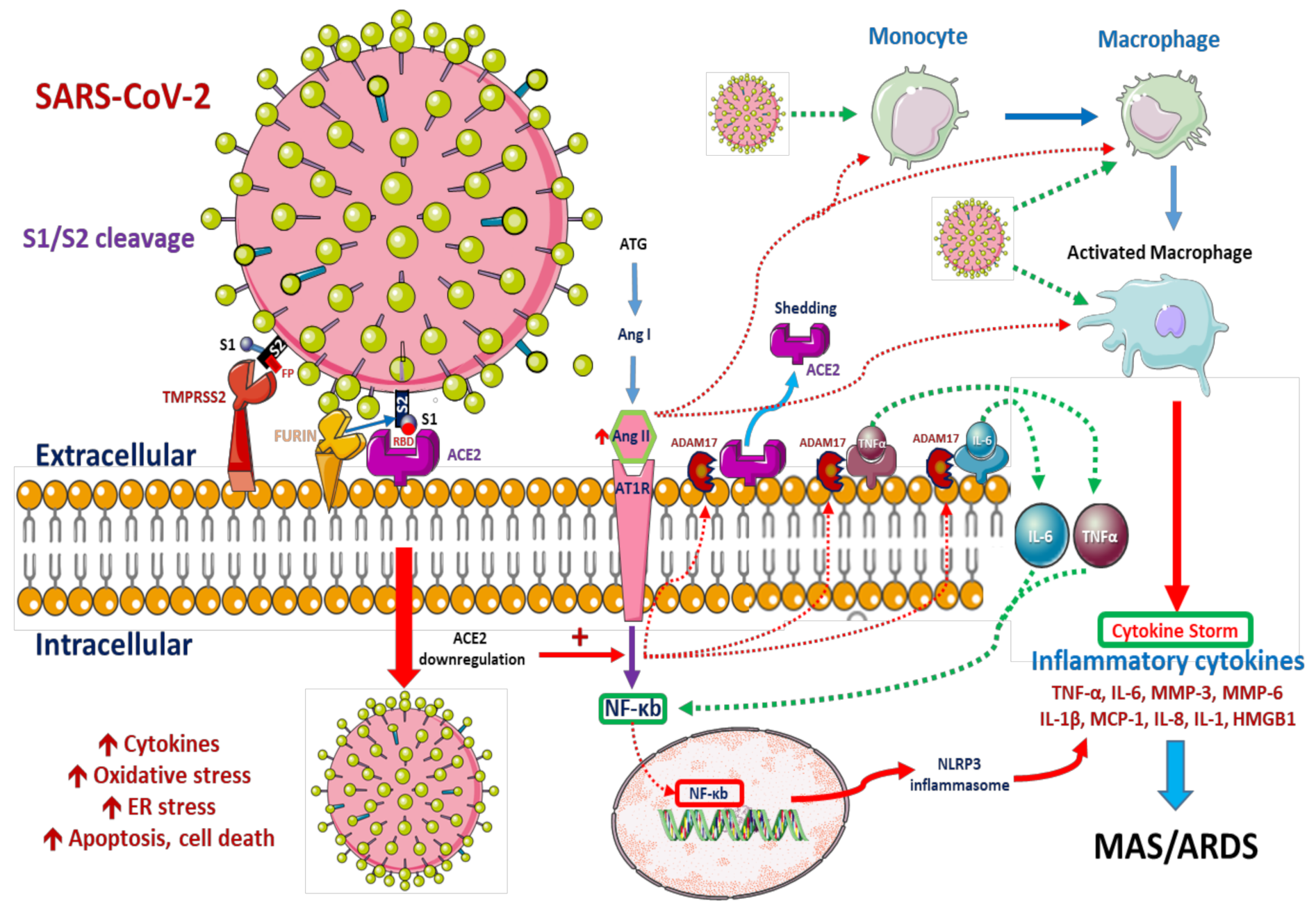{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. COVID-19 Pathophysiology
2.1. Viral Cell Entry
Role of Pulmonary Alveolar Type II Epithelial Cells and Macrophages in SARS-CoV-2 Infection
2.2. Innate Immune Responses
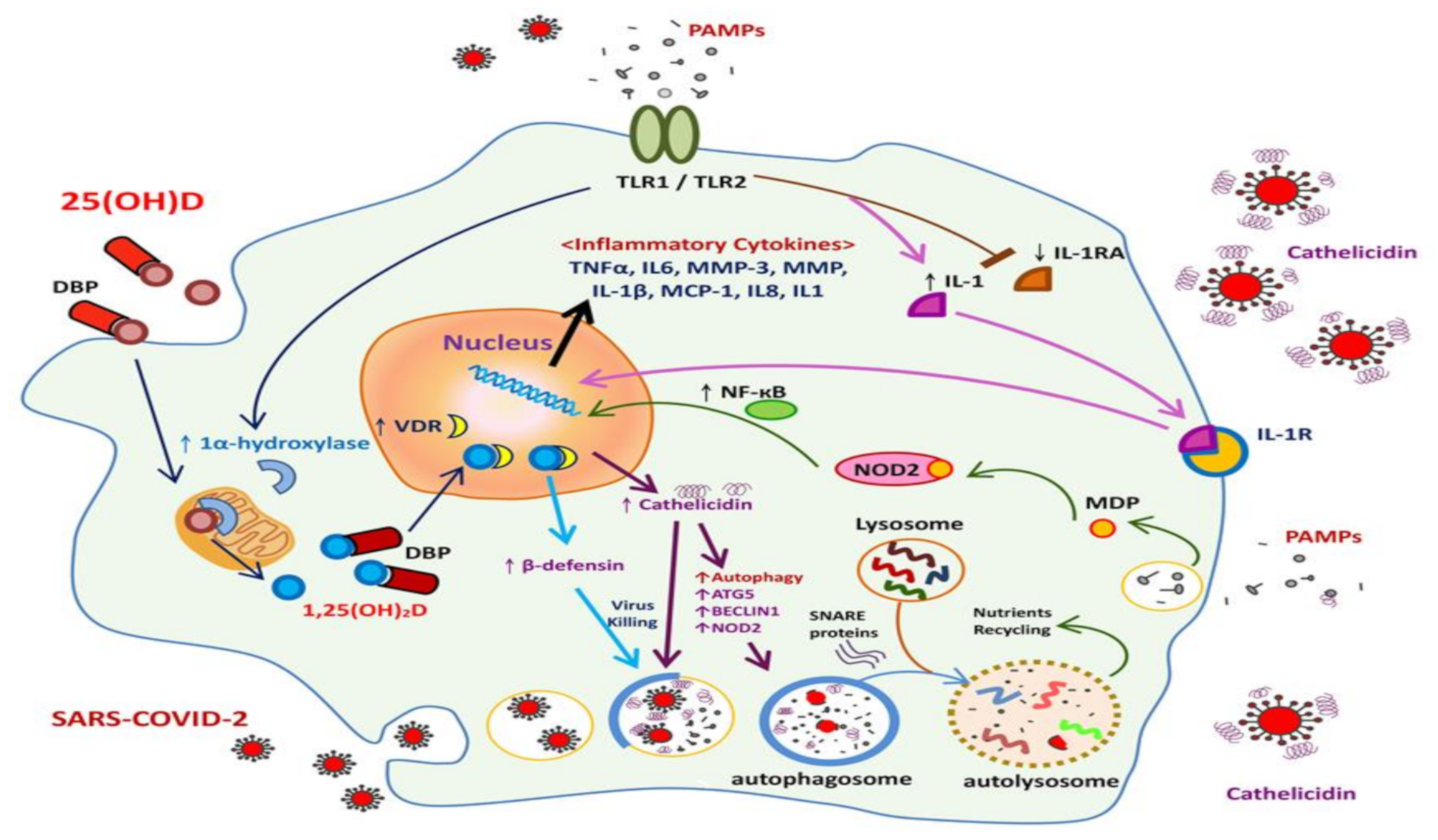
2.2.1. Local (Pulmonary) Innate Immune Responses to COVID-19 Infection
2.2.2. Systemic Innate Immune Responses against COVID-19 Infection
2.3. Adaptive Immune Response: T Cell Differentiation and Inflammatory Cytokines in COVID-19
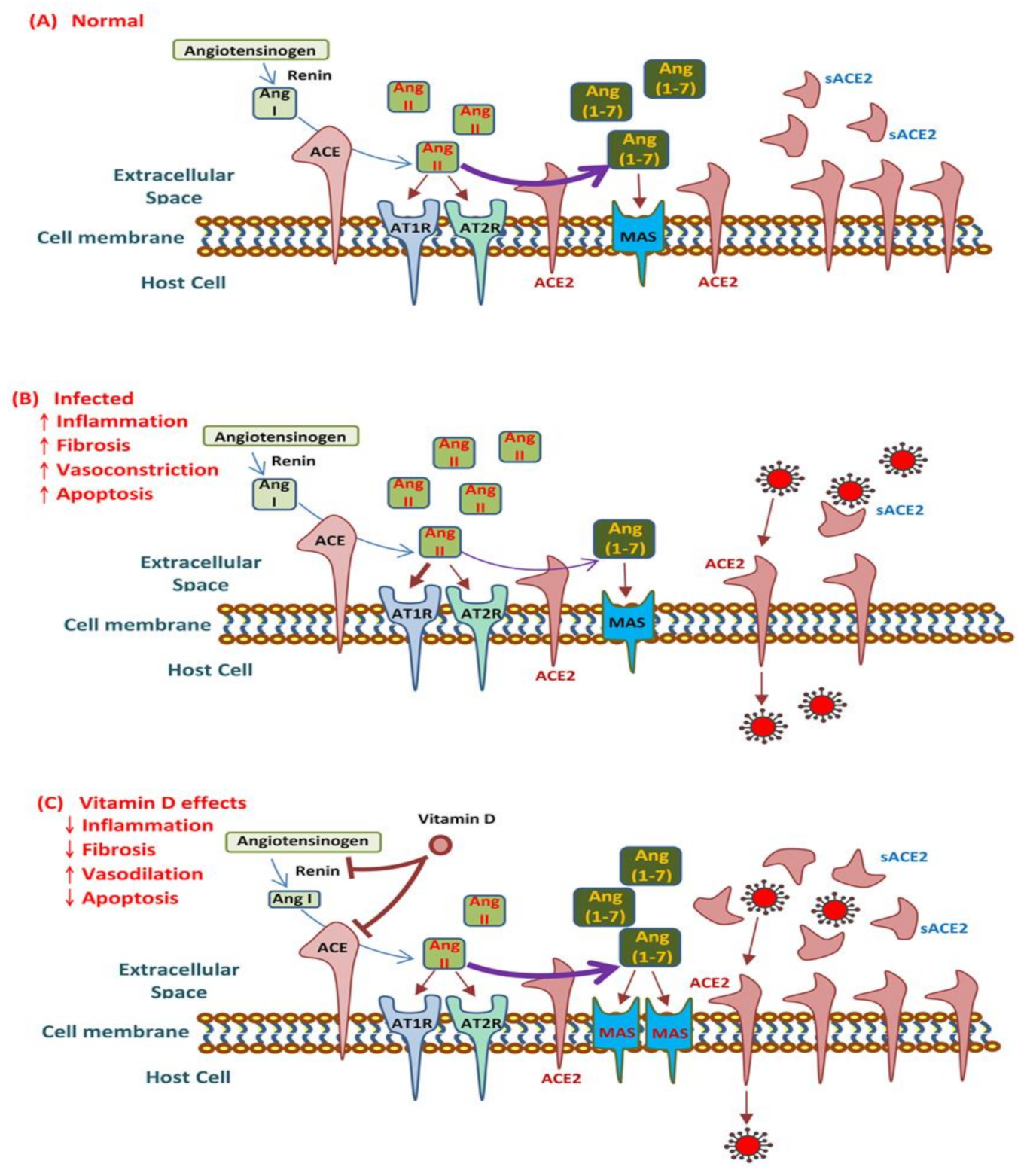
2.4. Renin–Angiotensin System and COVID-19
3. Role of Vitamin D in the COVID-19 Era
3.1. Antiviral Activity of Vitamin D and the Innate Immune Response
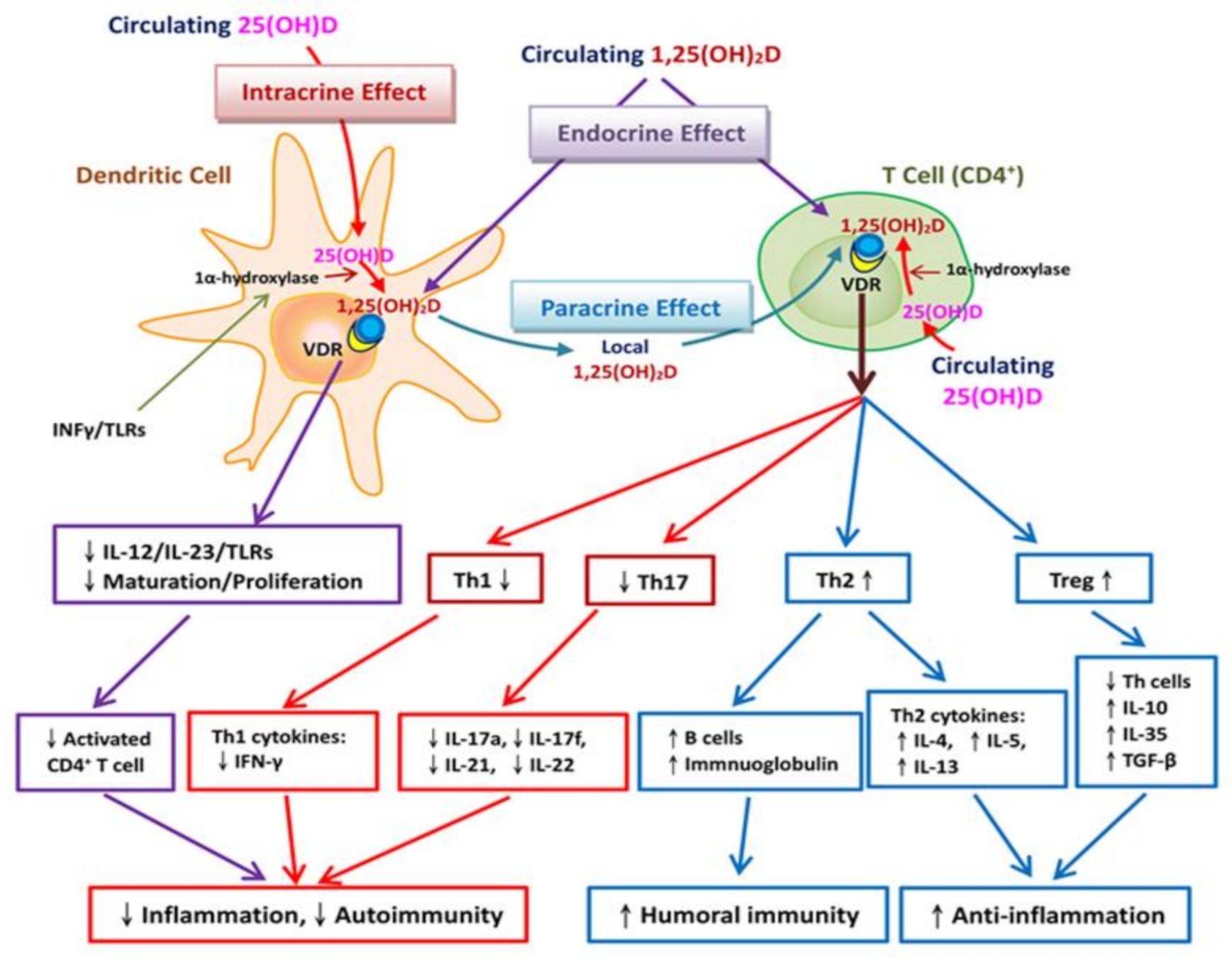
3.2. Vitamin D Regulates Adaptive Immunity
3.3. Vitamin D Modulates ACE2 and the RAS
4. Controversial Findings from Clinical Studies
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N. Role of vitamin D in preventing of COVID-19 infection, progression and severity. J. Infect. Public Health 2020, 10, 1373–1380. [Google Scholar] [CrossRef]
- Holick, M.F. The vitamin D deficiency pandemic: Approaches for diagnosis, treatment and prevention. Rev. Endocr. Metab. Disord. 2017, 18, 153–165. [Google Scholar] [CrossRef] [PubMed]
- D’Avolio, A.; Avataneo, V.; Manca, A.; Cusato, J.; De Nicolo, A.; Lucchini, R.; Keller, F.; Cantu, M. 25-Hydroxyvitamin D Concentrations Are Lower in Patients with Positive PCR for SARS-CoV-2. Nutrients 2020, 12, 1359. [Google Scholar] [CrossRef]
- Meltzer, D.O.; Best, T.J.; Zhang, H.; Vokes, T.; Arora, V.; Solway, J. Association of Vitamin D Status and Other Clinical Characteristics With COVID-19 Test Results. JAMA Netw. Open 2020, 3, e2019722. [Google Scholar] [CrossRef]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemb, P.; Bergman, P.; Camargo, C.A., Jr.; Cavalier, E.; Cormier, C.; Courbebaisse, M.; Hollis, B.; Joulia, F.; Minisola, S.; Pilz, S.; et al. Vitamin D deficiency and the COVID-19 pandemic. J. Glob. Antimicrob. Resist. 2020, 22, 133–134. [Google Scholar] [CrossRef]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “inflame-aging”. Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Kara, M.; Ekiz, T.; Ricci, V.; Kara, O.; Chang, K.V.; Ozcakar, L. ‘Scientific Strabismus’ or two related pandemics: Coronavirus disease and vitamin D deficiency. Br. J. Nutr. 2020, 124, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Bogliolo, L.; Lobascio, F.; Barichella, M.; Zecchinelli, A.L.; Pezzoli, G.; Caccialanza, R. Vitamin D supplementation and outcomes in coronavirus disease 2019 (COVID-19) patients from the outbreak area of Lombardy, Italy. Nutrition 2021, 82, 111055. [Google Scholar] [CrossRef]
- Ling, S.F.; Broad, E.; Murphy, R.; Pappachan, J.M.; Pardesi-Newton, S.; Kong, M.F.; Jude, E.B. High-Dose Cholecalciferol Booster Therapy is Associated with a Reduced Risk of Mortality in Patients with COVID-19: A Cross-Sectional Multi-Centre Observational Study. Nutrients 2020, 12, 3799. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, G.; Corvaisier, M.; Gautier, J.; Dubee, V.; Legrand, E.; Sacco, G.; Annweiler, C. Vitamin D Supplementation Associated to Better Survival in Hospitalized Frail Elderly COVID-19 Patients: The GERIA-COVID Quasi-Experimental Study. Nutrients 2020, 12, 3377. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Sims, A.C.; Baric, R.S.; Yount, B.; Burkett, S.E.; Collins, P.L.; Pickles, R.J. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: Role of ciliated cells in viral spread in the conducting airways of the lungs. J. Virol. 2005, 79, 15511–15524. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients with COVID-19 in Washington State. JAMA 2020. [Google Scholar] [CrossRef] [Green Version]
- Mossel, E.C.; Wang, J.; Jeffers, S.; Edeen, K.E.; Wang, S.; Cosgrove, G.P.; Funk, C.J.; Manzer, R.; Miura, T.A.; Pearson, L.D.; et al. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology 2008, 372, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wu, X.; Zeng, W.; Guo, D.; Fang, Z.; Chen, L.; Huang, H.; Li, C. Chest CT Findings in Patients With Coronavirus Disease 2019 and Its Relationship With Clinical Features. Investig. Radiol. 2020, 55, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Travanty, E.A.; Oko, L.; Edeen, K.; Berglund, A.; Wang, J.; Ito, Y.; Holmes, K.V.; Mason, R.J. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am. J. Respir. Cell Mol. Biol. 2013, 48, 742–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Zhang, J.; Ma, X.; Tan, J.; Chen, L.; Liu, S.; Xin, Y.; Zhuang, L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed. Pharmacother. 2020, 131, 110678. [Google Scholar] [CrossRef]
- Seyedpour, S.; Khodaei, B.; Loghman, A.H.; Seyedpour, N.; Kisomi, M.F.; Balibegloo, M.; Nezamabadi, S.S.; Gholami, B.; Saghazadeh, A.; Rezaei, N. Targeted therapy strategies against SARS-CoV-2 cell entry mechanisms: A systematic review of in vitro and in vivo studies. J. Cell Physiol. 2021, 236, 2364–2392. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, A.; Scavone, C.; Rafaniello, C.; Ferrajolo, C.; Racagni, G.; Berrino, L.; Paolisso, G.; Rossi, F.; Capuano, A. Renin-Angiotensin System and Coronavirus Disease 2019: A Narrative Review. Front. Cardiovasc. Med. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1–7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Banu, N.; Panikar, S.S.; Leal, L.R.; Leal, A.R. Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: Therapeutic implications. Life Sci. 2020, 256, 117905. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Smith, G.J.; Zhang, J.X.; Peiris, J.S.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [Green Version]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.T.; Jia, N.; Zhang, Y.W.; Shum, M.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Lan, L.; Xu, D.; Ye, G.; Xia, C.; Wang, S.; Li, Y.; Xu, H. Positive RT-PCR Test Results in Patients Recovered From COVID-19. JAMA 2020, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abassi, Z.; Knaney, Y.; Karram, T.; Heyman, S.N. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 2020, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Hill, T.; Li, K.; Peters, C.J.; Tseng, C.T. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 2009, 83, 3039–3048. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890.e2. [Google Scholar] [CrossRef]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Ganz, T. The role of antimicrobial peptides in innate immunity. Integr. Comp. Biol. 2003, 43, 300–304. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.L.; McDermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: Biological and therapeutic considerations. Exp. Dermatol. 2016, 25, 167–173. [Google Scholar] [CrossRef]
- Weidmann, J.; Craik, D.J. Discovery, structure, function, and applications of cyclotides: Circular proteins from plants. J. Exp. Bot. 2016, 67, 4801–4812. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, G.; Gabrani, R. Antiviral Peptides: Identification and Validation. Int. J. Pept. Res. Ther. 2020, 1–20. [Google Scholar] [CrossRef]
- Ahmed, A.; Siman-Tov, G.; Keck, F.; Kortchak, S.; Bakovic, A.; Risner, K.; Lu, T.K.; Bhalla, N.; de la Fuente-Nunez, C.; Narayanan, A. Human cathelicidin peptide LL-37 as a therapeutic antiviral targeting Venezuelan equine encephalitis virus infections. Antiviral. Res. 2019, 164, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Klotman, M.E.; Chang, T.L. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Pachon-Ibanez, M.E.; Smani, Y.; Pachon, J.; Sanchez-Cespedes, J. Perspectives for clinical use of engineered human host defense antimicrobial peptides. FEMS Microbiol. Rev. 2017, 41, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Sanchez, J.I.; Dominguez-Martinez, D.A.; Olivar-Espinosa, N.; Garcia-Perez, B.E.; Lorono-Pino, M.A.; Luna-Herrera, J.; Salazar, M.I. Expression of Antimicrobial Peptides in Human Monocytic Cells and Neutrophils in Response to Dengue Virus Type 2. Intervirology 2016, 59, 8–19. [Google Scholar] [CrossRef]
- Findlay, F.; Proudfoot, L.; Stevens, C.; Barlow, P.G. Cationic host defense peptides; novel antimicrobial therapeutics against Category A pathogens and emerging infections. Pathog. Glob. Health 2016, 110, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Misawa, N.; Takeuchi, J.S.; Kobayashi, T.; Izumi, T.; Aso, H.; Nagaoka, S.; Yamamoto, K.; Kimura, I.; Konno, Y.; et al. Experimental Adaptive Evolution of Simian Immunodeficiency Virus SIVcpz to Pandemic Human Immunodeficiency Virus Type 1 by Using a Humanized Mouse Model. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lau, Y.F.; Lamirande, E.W.; Paddock, C.D.; Bartlett, J.H.; Zaki, S.R.; Subbarao, K. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 2010, 84, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef]
- Liu, T.; Zeng, G.; Tao, H.; Shi, Y.; Group, C.-i.C.P.R.; Wang, T.; Liu, T.; Guo, F.; Zhou, F.; Wang, X. Low prevalence of IgG antibodies to SARS-CoV-2 in cancer patients with COVID-19. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- Long, Q.X.; Tang, X.J.; Shi, Q.L.; Li, Q.; Deng, H.J.; Yuan, J.; Hu, J.L.; Xu, W.; Zhang, Y.; Lv, F.J.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.G.; Mendoza, A.; Hemmers, S.; Moltedo, B.; Niec, R.E.; Schizas, M.; Hoyos, B.E.; Putintseva, E.V.; Chaudhry, A.; Dikiy, S.; et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 2017, 546, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.L.; Wang, W.J.; Chen, D.X.; Xu, B. Dysregulation of the immune response affects the outcome of critical COVID-19 patients. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Remy, K.E.; Mazer, M.; Striker, D.A.; Ellebedy, A.H.; Walton, A.H.; Unsinger, J.; Blood, T.M.; Mudd, P.A.; Yi, D.J.; Mannion, D.A.; et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, J.; Bhattachariya, A.; Alajbegovic, A.; Rippe, C.; Ekman, M.; Dahan, D.; Hien, T.T.; Boettger, T.; Braun, T.; Sward, K.; et al. Loss of Vascular Myogenic Tone in miR-143/145 Knockout Mice Is Associated With Hypertension-Induced Vascular Lesions in Small Mesenteric Arteries. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 414–424. [Google Scholar] [CrossRef]
- Brojakowska, A.; Narula, J.; Shimony, R.; Bander, J. Clinical Implications of SARS-CoV-2 Interaction With Renin Angiotensin System: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 3085–3095. [Google Scholar] [CrossRef]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.C.; Pickering, R.J.; Tsorotes, D.; Koitka, A.; Sheehy, K.; Bernardi, S.; Toffoli, B.; Nguyen-Huu, T.P.; Head, G.A.; Fu, Y.; et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 2010, 107, 888–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Sfera, A.; Osorio, C.; Jafri, N.; Diaz, E.L.; Campo Maldonado, J.E. Intoxication With Endogenous Angiotensin II: A COVID-19 Hypothesis. Front. Immunol. 2020, 11, 1472. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Cingolani, O.; Lakatta, E.G. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights From Cardiovascular Aging Science. JAMA Cardiol. 2020, 5, 747–748. [Google Scholar] [CrossRef] [Green Version]
- Gwyer Findlay, E.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Zdrenghea, M.T.; Makrinioti, H.; Bagacean, C.; Bush, A.; Johnston, S.L.; Stanciu, L.A. Vitamin D modulation of innate immune responses to respiratory viral infections. Rev. Med. Virol. 2017, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Zheng, C.M.; Lu, C.L.; Lin, Y.F.; Shyu, J.F.; Wu, C.C.; Lu, K.C. Vitamin D and immune function in chronic kidney disease. Clin. Chim. Acta 2015, 450, 135–144. [Google Scholar] [CrossRef]
- Matsumura, T.; Sugiyama, N.; Murayama, A.; Yamada, N.; Shiina, M.; Asabe, S.; Wakita, T.; Imawari, M.; Kato, T. Antimicrobial peptide LL-37 attenuates infection of hepatitis C virus. Hepatol. Res. 2016, 46, 924–932. [Google Scholar] [CrossRef]
- Su, D.; Nie, Y.; Zhu, A.; Chen, Z.; Wu, P.; Zhang, L.; Luo, M.; Sun, Q.; Cai, L.; Lai, Y.; et al. Vitamin D Signaling through Induction of Paneth Cell Defensins Maintains Gut Microbiota and Improves Metabolic Disorders and Hepatic Steatosis in Animal Models. Front. Physiol. 2016, 7, 498. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Q.; Hou, Y.C.; Zheng, C.M.; Lu, C.L.; Liu, W.C.; Wu, C.C.; Huang, M.T.; Lin, Y.F.; Lu, K.C. Cholecalciferol Additively Reduces Serum Parathyroid Hormone and Increases Vitamin D and Cathelicidin Levels in Paricalcitol-Treated Secondary Hyperparathyroid Hemodialysis Patients. Nutrients 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, M.; Merugu, G.P.; Konala, V.M.; Sangani, V.; Kondakindi, H.; Pokal, M.; Gayam, V.; Adapa, S.; Naramala, S.; Malayala, S.V. Back to basics: Review on vitamin D and respiratory viral infections including COVID-19. J. Community Hosp. Intern. Med. Perspect. 2020, 10, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D receptor inhibits nuclear factor kappaB activation by interacting with IkappaB kinase beta protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef] [Green Version]
- Hansdottir, S.; Monick, M.M.; Hinde, S.L.; Lovan, N.; Look, D.C.; Hunninghake, G.W. Respiratory epithelial cells convert inactive vitamin D to its active form: Potential effects on host defense. J. Immunol. 2008, 181, 7090–7099. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, R.L.; Sternberg, M.R.; Looker, A.C.; Yetley, E.A.; Lacher, D.A.; Sempos, C.T.; Taylor, C.L.; Durazo-Arvizu, R.A.; Maw, K.L.; Chaudhary-Webb, M.; et al. National Estimates of Serum Total 25-Hydroxyvitamin D and Metabolite Concentrations Measured by Liquid Chromatography-Tandem Mass Spectrometry in the US Population during 2007–2010. J. Nutr. 2016, 146, 1051–1061. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.; Zheng, J.Q.; Wu, C.C.; Hou, Y.C.; Liu, W.C.; Lu, C.L.; Zheng, C.M.; Lu, K.C.; Chao, Y.C. Perspective Adjunctive Therapies for COVID-19: Beyond Antiviral Therapy. Int. J. Med. Sci. 2021, 18, 314–324. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Chen, J.; Luo, Q.; Zhang, Q.; Zhang, H. Vitamin D alleviates lipopolysaccharideinduced acute lung injury via regulation of the reninangiotensin system. Mol. Med. Rep. 2017, 16, 7432–7438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunutsor, S.K.; Apekey, T.A.; Steur, M. Vitamin D and risk of future hypertension: Meta-analysis of 283,537 participants. Eur. J. Epidemiol. 2013, 28, 205–221. [Google Scholar] [CrossRef] [PubMed]
- McMullan, C.J.; Borgi, L.; Curhan, G.C.; Fisher, N.; Forman, J.P. The effect of vitamin D on renin-angiotensin system activation and blood pressure: A randomized control trial. J. Hypertens. 2017, 35, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, D.; Caccamo, D.; Lucisano, S.; Buemi, M.; Sebekova, K.; Teta, D.; De Nicola, L. Interplay of vitamin D, erythropoiesis, and the renin-angiotensin system. BioMed Res. Int. 2015, 2015, 145828. [Google Scholar] [CrossRef] [Green Version]
- Tomaschitz, A.; Pilz, S.; Ritz, E.; Grammer, T.; Drechsler, C.; Boehm, B.O.; Marz, W. Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin. Chim. Acta 2010, 411, 1354–1360. [Google Scholar] [CrossRef]
- Cure, E.; Cumhur Cure, M. Comment on “Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19”. J. Med. Virol. 2020, 14. [Google Scholar] [CrossRef] [Green Version]
- Hanff, T.C.; Harhay, M.O.; Brown, T.S.; Cohen, J.B.; Mohareb, A.M. Is There an Association Between COVID-19 Mortality and the Renin-Angiotensin System-a Call for Epidemiologic Investigations. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Uri, K.; Fagyas, M.; Manyine Siket, I.; Kertesz, A.; Csanadi, Z.; Sandorfi, G.; Clemens, M.; Fedor, R.; Papp, Z.; Edes, I.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE 2014, 9, e87845. [Google Scholar] [CrossRef]
- Soro-Paavonen, A.; Gordin, D.; Forsblom, C.; Rosengard-Barlund, M.; Waden, J.; Thorn, L.; Sandholm, N.; Thomas, M.C.; Groop, P.H.; FinnDiane Study, G. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J. Hypertens. 2012, 30, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, 84–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Gao, P.; Zhao, T.; He, L.; Li, M.; Li, Y.; Shui, H.; Wu, X. Calcitriol regulates angiotensin-converting enzyme and angiotensin converting-enzyme 2 in diabetic kidney disease. Mol. Biol. Rep. 2016, 43, 397–406. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, E.K.; Thenappan, T.; Bhargava, M.; Chen, Y. Does vitamin D deficiency increase the severity of COVID-19? Clin. Med. (Lond) 2020, 20, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Hastie, C.E.; Mackay, D.F.; Ho, F.; Celis-Morales, C.A.; Katikireddi, S.V.; Niedzwiedz, C.L.; Jani, B.D.; Welsh, P.; Mair, F.S.; Gray, S.R.; et al. Vitamin D concentrations and COVID-19 infection in UK Biobank. Diabetes Metab. Syndr. 2020, 14, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Chandran, M.; Chan Maung, A.; Mithal, A.; Parameswaran, R. Vitamin D in COVID-19: Dousing the fire or averting the storm?—A perspective from the Asia-Pacific. Osteoporos. Sarcopenia 2020. [Google Scholar] [CrossRef]
- Mercola, J.; Grant, W.B.; Wagner, C.L. Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity. Nutrients 2020, 12, 3361. [Google Scholar] [CrossRef]
- Hill, A.B. The environment and disease: Association or causation? J. R. Soc. Med. 2015, 108, 32–37. [Google Scholar] [CrossRef]
- Mitchell, F. Vitamin-D and COVID-19: Do deficient risk a poorer outcome? Lancet Diabetes Endocrinol. 2020, 8, 570. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, M.-Y.; Liu, W.-C.; Zheng, J.-Q.; Lu, C.-L.; Hou, Y.-C.; Zheng, C.-M.; Song, J.-Y.; Lu, K.-C.; Chao, Y.-C. Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. Int. J. Mol. Sci. 2021, 22, 5251. https://doi.org/10.3390/ijms22105251
Peng M-Y, Liu W-C, Zheng J-Q, Lu C-L, Hou Y-C, Zheng C-M, Song J-Y, Lu K-C, Chao Y-C. Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. International Journal of Molecular Sciences. 2021; 22(10):5251. https://doi.org/10.3390/ijms22105251
Chicago/Turabian StylePeng, Ming-Yieh, Wen-Chih Liu, Jing-Quan Zheng, Chien-Lin Lu, Yi-Chou Hou, Cai-Mei Zheng, Jenn-Yeu Song, Kuo-Cheng Lu, and You-Chen Chao. 2021. "Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D" International Journal of Molecular Sciences 22, no. 10: 5251. https://doi.org/10.3390/ijms22105251
APA StylePeng, M. -Y., Liu, W. -C., Zheng, J. -Q., Lu, C. -L., Hou, Y. -C., Zheng, C. -M., Song, J. -Y., Lu, K. -C., & Chao, Y. -C. (2021). Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. International Journal of Molecular Sciences, 22(10), 5251. https://doi.org/10.3390/ijms22105251