
Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies



Abstract
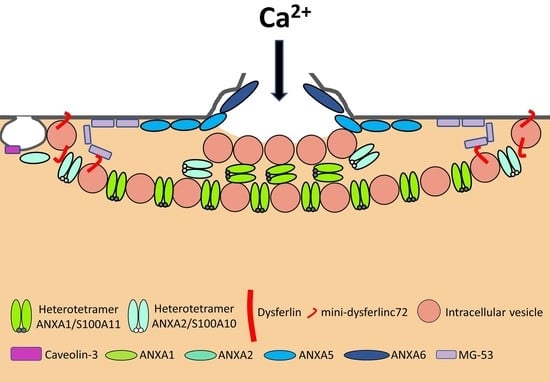
:
1. Introduction
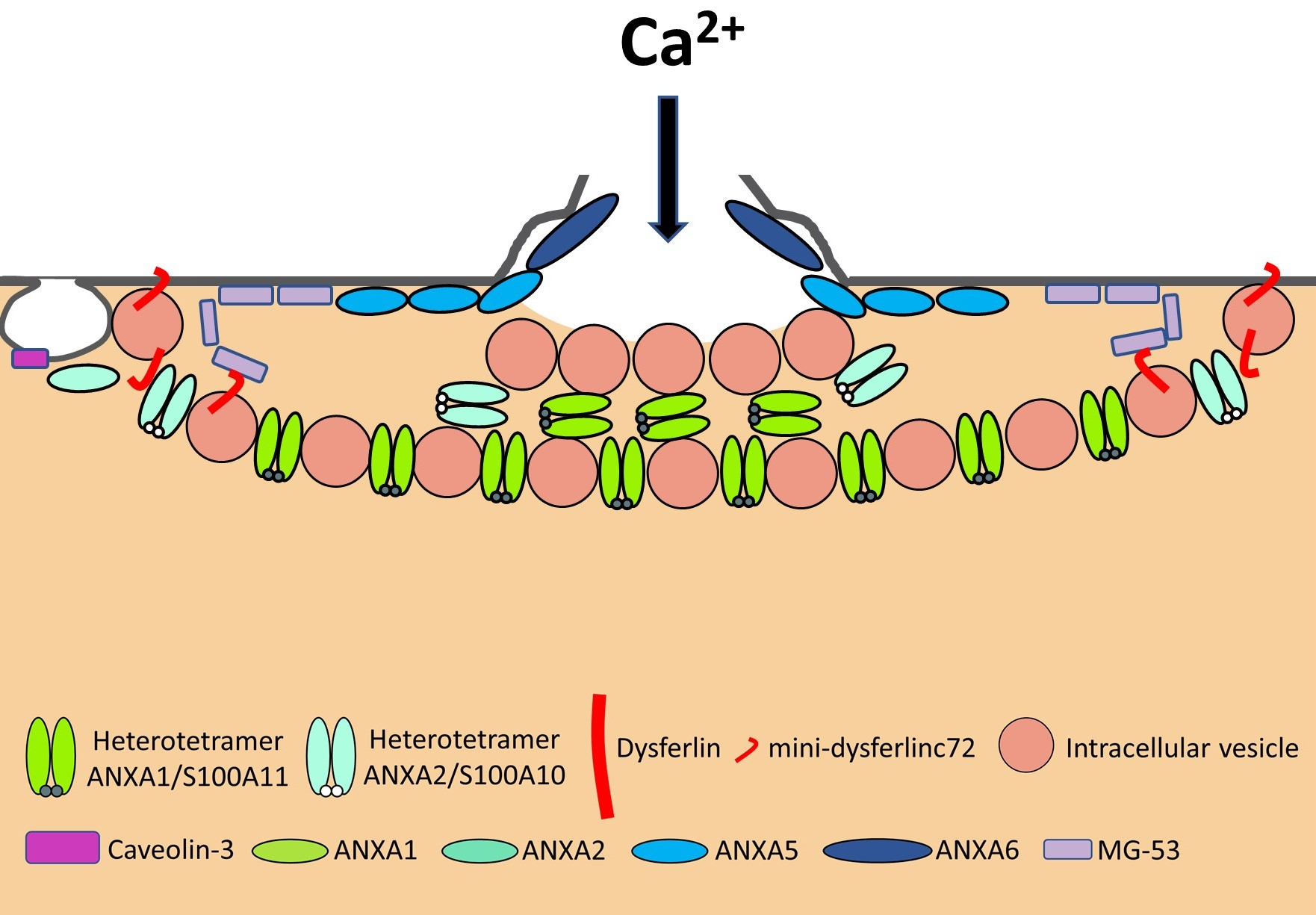
2. Anatomy of Skeletal Muscle
3. The Sarcolemma Repair Machinery
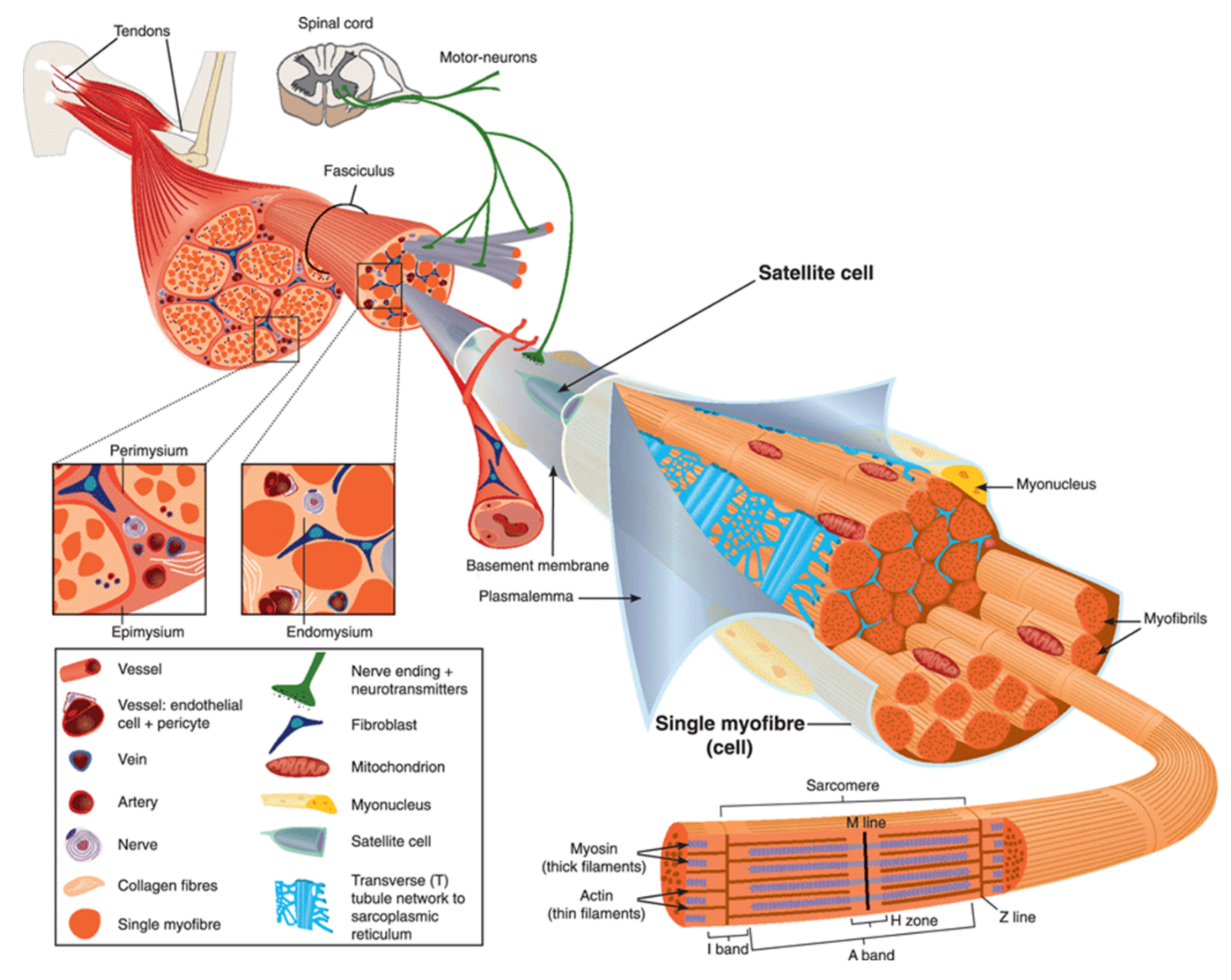
3.1. The “Lipid Patch” Mechanism
3.2. The Sarcolemma Repair Proteins
3.2.1. Dysferlin
3.2.2. Caveolin-3
3.2.3. Anoctamin-5
3.2.4. MG53
3.2.5. Calpains
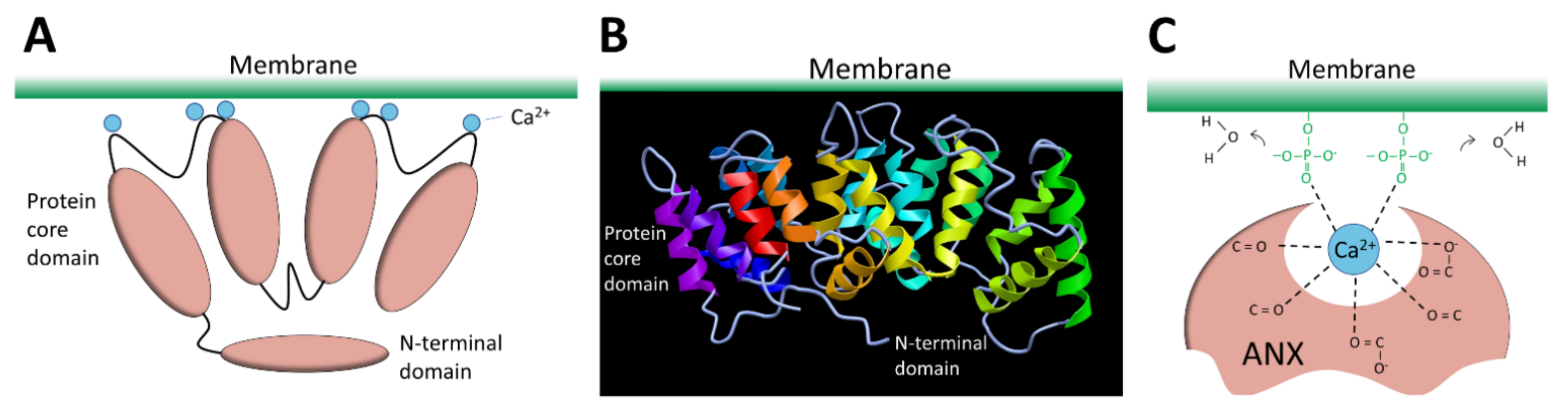
3.2.6. Annexins
ANXA1 et A2
ANXA4
ANXA5
ANXA6
ANXA7
4. Aetiology and Nomenclature of Muscular Dystrophies
5. Membrane Repair and Muscular Dystrophies
5.1. LGMDR2 Dysferlin-Related (LGMD2B)
5.2. LGMDD4 Calpain3-Related (LGMD1I) and LGMDR1 Calpain3-Related (LGMD2A)
5.3. LGMDR12 Anoctamin5-Related (LGMD2L)
5.4. Rippling Muscle Disease Caveolin-3 Related (LGMD1C)
5.5. Duchenne Muscular Dystrophy Dystrophin-Related
6. ANXA and Muscular Dystrophies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef]
- Zhen, Y.; Radulovic, M.; Vietri, M.; Stenmark, H. Sealing holes in cellular membranes. EMBO J. 2021, 40, e106922. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Ito, S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology 1989, 96, 1238–1248. [Google Scholar] [CrossRef]
- Yu, Q.C.; McNeil, P.L. Transient disruptions of aortic endothelial cell plasma membranes. Am. J. Pathol. 1992, 141, 1349–1360. [Google Scholar]
- McNeil, P.L.; Khakee, R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992, 140, 1097–1109. [Google Scholar]
- Clarke, M.S.; Caldwell, R.W.; Chiao, H.; Miyake, K.; McNeil, P.L. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ. Res. 1995, 76, 927–934. [Google Scholar] [CrossRef]
- Horn, A.; Jaiswal, J.K. Cellular mechanisms and signals that coordinate plasma membrane repair. Cell. Mol. Life Sci. 2018, 75, 3751–3770. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Andoni Urtizberea, J.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Minetti, C.; Sotgia, F.; Bruno, C.; Scartezzini, P.; Broda, P.; Bado, M.; Masetti, E.; Mazzocco, M.; Egeo, A.; Donati, M.A.; et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998, 18, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.A.; Johnson, R.W.; Whitlock, J.M.; Pozsgai, E.R.; Heller, K.N.; Grose, W.E.; Arnold, W.D.; Sahenk, Z.; Hartzell, H.C.; Rodino-Klapac, L.R. Defective membrane fusion and repair in Anoctamin5 -deficient muscular dystrophy. Hum. Mol. Genet. 2016, 25, 1900–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Xu, L.; Lau, Y.S.; Gao, Y.; Moore, S.A.; Han, R. A novel ANO5 splicing variant in a LGMD2L patient leads to production of a truncated aggregation-prone Ano5 peptide. J. Pathol. Clin. Res. 2018, 4, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.S.F.; Khakee, R.; McNeil, P.L. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J. Cell Sci. 1993, 106, 121–133. [Google Scholar] [CrossRef]
- Tajbakhsh, S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J. Intern. Med. 2009, 266, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Rudnicki, M.A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 127–133. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Ann. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [Green Version]
- Middel, V.; Zhou, L.; Takamiya, M.; Beil, T.; Shahid, M.; Roostalu, U.; Grabher, C.; Rastegar, S.; Reischl, M.; Nienhaus, G.U.; et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat. Commun. 2016, 7, 12875. [Google Scholar] [CrossRef]
- Draeger, A.; Schoenauer, R.; Atanassoff, A.P.; Wolfmeier, H.; Babiychuk, E.B. Dealing with damage: Plasma membrane repair mechanisms. Biochimie 2014, 107, 66–72. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Perez, F. Plasma membrane repair: The adaptable cell life-insurance. Curr. Opin. Cell Biol. 2017, 47, 99–107. [Google Scholar] [CrossRef]
- Carmeille, R.; Bouvet, F.; Tan, S.; Croissant, C.; Gounou, C.; Mamchaoui, K.; Mouly, V.; Brisson, A.R.; Bouter, A. Membrane repair of human skeletal muscle cells requires Annexin-A5. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2267–2279. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Bouvet, F.; Tan, S.; Bouter, A. Imaging membrane repair in single cells using correlative light and electron microscopy. Curr. Protoc. Cell Biol. 2018, 81, e55. [Google Scholar] [CrossRef]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Annexin-A6 in membrane repair of human skeletal muscle cell: A role in the cap subdomain. Cells 2020, 9, 1742. [Google Scholar] [CrossRef]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef] [Green Version]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Miyake, K.; Vogel, S.S. The endomembrane requirement for cell surface repair. Proc. Natl. Acad. Sci. USA 2003, 100, 4592–4597. [Google Scholar] [CrossRef] [Green Version]
- Eddleman, C.S.; Ballinger, M.L.; Smyers, M.E.; Fishman, H.M.; Bittner, G.D. Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury. J. Neurosci. 1998, 18, 4029–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgonovo, B.; Cocucci, E.; Racchetti, G.; Podini, P.; Bachi, A.; Meldolesi, J. Regulated exocytosis: A novel, widely expressed system. Nat. Cell Biol. 2002, 4, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Medikayala, S.; Defour, A.; Rayavarapu, S.; Brown, K.J.; Hathout, Y.; Jaiswal, J.K. Use of quantitative membrane proteomics identifies a novel role of mitochondria in healing injured muscles. J. Biol. Chem. 2012, 287, 30455–30467. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Blazek, A.D.; Paleo, B.J.; Weisleder, N. Plasma membrane repair: A central process for maintaining cellular homeostasis. Physiology 2015, 30, 438–448. [Google Scholar] [CrossRef]
- Barthélémy, F.; Defour, A.; Lévy, N.; Krahn, M.; Bartoli, M. Muscle Cells Fix Breaches by Orchestrating a Membrane Repair Ballet; IOS Press: Amsterdam, The Netherlands, 2018; Volume 5, pp. 21–28. [Google Scholar]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 mediates dysferlin accumulation and muscle cell membrane repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [Green Version]
- Koerdt, S.N.; Gerke, V. Annexin A2 is involved in Ca2+-dependent plasma membrane repair in primary human endothelial cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1046–1053. [Google Scholar] [CrossRef]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [Green Version]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; D’Estaintot, B.L.B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Roostalu, U.; Strähle, U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Fallon, K.S.; Oosterbaan, C.C.; Bogdanovic, E.; Warner, J.L.; Sell, J.J.; Page, P.G.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. Recombinant annexin A6 promotes membrane repair and protects against muscle injury. J. Clin. Investig. 2019, 129, 4657–4670. [Google Scholar] [CrossRef] [PubMed]
- Sønder, S.L.; Boye, T.L.; Tölle, R.; Dengjel, J.; Maeda, K.; Jäättelä, M.; Simonsen, A.C.; Jaiswal, J.K.; Nylandsted, J. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci. Rep. 2019, 9, 6726. [Google Scholar] [CrossRef] [PubMed]
- Selbert, S.; Fischer, P.; Menke, A.; Jockusch, H.; Pongratz, D.; Noegel, A.A. Annexin VII relocalization as a result of dystrophin deficiency. Exp. Cell Res. 1996, 222, 199–208. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Marlow, G.; Summerill, G.; Mahjneh, I.; Mueller, S.; Hill, M.; Miyake, K.; Haase, H.; Anderson, L.V.B.; Richard, I.; et al. Patients with a non-dysferlin miyoshi myopathy have a novel membrane repair defect. Traffic 2007, 8, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, V.; Marlow, G.; Boycott, K.M.; Saleki, K.; Inoue, H.; Kroon, J.; Itakura, M.; Robitaille, Y.; Parent, L.; Baas, F.; et al. recessive mutations in the putative calcium-activated chloride channel anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 2010, 86, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Monjaret, F.; Suel-Petat, L.; Bourg-Alibert, N.; Vihola, A.; Marchand, S.; Roudaut, C.; Gicquel, E.; Udd, B.; Richard, I.; Charton, K. The phenotype of dysferlin-deficient mice is not rescued by adeno-associated virus-mediated transfer of anoctamin 5. Hum. Gene Ther. Clin. Dev. 2013, 24, 65–76. [Google Scholar] [CrossRef]
- Chandra, G.; Defour, A.; Mamchoui, K.; Pandey, K.; Mishra, S.; Mouly, V.; Sreetama, S.C.; Mahad Ahmad, M.; Mahjneh, I.; Morizono, H.; et al. Dysregulated calcium homeostasis prevents plasma membrane repair in Anoctamin 5/TMEM16E-deficient patient muscle cells. Cell Death Discov. 2019, 5, 118. [Google Scholar] [CrossRef] [Green Version]
- Foltz, S.J.; Cui, Y.Y.; Choo, H.J.; Hartzell, H.C. ANO5 ensures trafficking of annexins in wounded myofibers. J. Cell Biol. 2021, 220, e202007059. [Google Scholar] [CrossRef] [PubMed]
- Lek, A.; Evesson, F.J.; Lemckert, F.A.; Redpath, G.M.I.; Lueders, A.-K.; Turnbull, L.; Whitchurch, C.B.; North, K.N.; Cooper, S.T. Calpains, cleaved Mini-DysferlinC72, and L-Type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 2013, 33, 5085–5094. [Google Scholar] [CrossRef] [Green Version]
- Mellgren, R.L.; Miyake, K.; Kramerova, I.; Spencer, M.J.; Bourg, N.; Bartoli, M.; Richard, I.; Greer, P.A.; McNeil, P.L. Calcium-dependent plasma membrane repair requires m- or μ-calpain, but not calpain-3, the proteasome, or caspases. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1886–1893. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; De Morrée, A.; Van Remoortere, A.; Bushby, K.; Frants, R.R.; Dunnen, J.T.; Van der Maarel, S.M. Calpain 3 is a modulator of the dysferlin protein complex in skeletal muscle. Hum. Mol. Genet. 2008, 17, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellgren, R.L.; Zhang, W.; Miyake, K.; McNeil, P.L. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J. Biol. Chem. 2007, 282, 2567–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, C.; Hayashi, Y.K.; Ogawa, M.; Aoki, M.; Murayama, K.; Nishino, I.; Nonaka, I.; Arahata, K.; Brown, R.H., Jr. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum. Mol. Genet. 2001, 10, 1761–1766. [Google Scholar] [CrossRef]
- Parton, R.G.; Del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Lisanti, M.P. Caveolins and caveolae: Molecular and functional relationships. Exp. Cell Res. 2001, 271, 36–44. [Google Scholar] [CrossRef]
- Sinha, B.; Köster, D.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Vedie, B.; Johannes, L.; et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011, 144, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Cai, C.; Weisleder, N.; Ko, J.-K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998, 20, 37–42. [Google Scholar] [CrossRef]
- Marty, N.J.; Holman, C.L.; Abdullah, N.; Johnson, C.P. The C2 domains of otoferlin, dysferlin, and myoferlin alter the packing of lipid bilayers. Biochemistry 2013, 52, 5585–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redpath, G.M.I.; Woolger, N.; Piper, A.K.; Lemckert, F.A.; Lek, A.; Greer, P.A.; North, K.N.; Cooper, S.T. Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repair. Mol. Biol. Cell 2014, 25, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; Head, S.I. Membrane injury and repair in the muscular dystrophies. Neuroscientist 2015, 21, 653–668. [Google Scholar] [CrossRef]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.-K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef] [Green Version]
- Piper, A.-K.; Sophocleous, R.A.; Ross, S.E.; Evesson, F.J.; Saleh, O.; Bournazos, A.; Yasa, J.; Reed, C.; Woolger, N.; Sluyter, R.; et al. Loss of calpains-1 and -2 prevents repair of plasma membrane scrape injuries, but not small pores, and induces a severe muscular dystrophy. Am. J. Physiol. Physiol. 2020, 318, C1226–C1237. [Google Scholar] [CrossRef]
- Jahnke, V.E.; Peterson, J.M.; Van Der Meulen, J.H.; Boehler, J.; Uaesoontrachoon, K.; Johnston, H.K.; Defour, A.; Phadke, A.; Yu, Q.; Jaiswal, J.K.; et al. Mitochondrial dysfunction and consequences in calpain-3-deficient muscle. Skelet. Muscle 2020, 10, 37. [Google Scholar] [CrossRef]
- Blandin, G.; Beroud, C.; Labelle, V.; Nguyen, K.; Wein, N.; Hamroun, D.; Williams, B.; Monnier, N.; Rufibach, L.E.; Urtizberea, J.A.; et al. UMD-DYSF, a novel locus specific database for the compilation and interactive analysis of mutations in the dysferlin gene. Hum. Mutat. 2012, 33, E2317–E2331. [Google Scholar] [CrossRef]
- Krahn, M.; Wein, N.; Bartoli, M.; Lostal, W.; Courrier, S.; Bourg-Alibert, N.; Nguyen, K.; Vial, C.; Streichenberger, N.; Labelle, V.; et al. A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathy. Sci. Transl. Med. 2010, 2, 50ra69. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.; Sarkozy, A.; Muelas, N.; Koehler, K.; Huebner, A.; Hudson, G.; Chinnery, P.F.; Barresi, R.; Eagle, M.; Polvikoski, T.; et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 2011, 134, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC International Workshop: Limb Girdle Muscular Dystrophies—Nomenclature and Reformed Classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazzerro, E.; Sotgia, F.; Bruno, C.; Lisanti, M.P.; Minetti, C. Caveolinopathies: From the biology of caveolin-3 to human diseases. Eur. J. Hum. Genet. 2010, 18, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Fee, D.B.; So, Y.T.; Barraza, C.; Figueroa, K.P.; Pulst, S.M. Phenotypic variability associated with Arg26Gln mutation in caveolin3. Muscle Nerve 2004, 30, 375–378. [Google Scholar] [CrossRef]
- Galbiati, F.; Volonte, D.; Minetti, C.; Chu, J.B.; Lisanti, M.P. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J. Biol. Chem. 1999, 274, 25632–25641. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, F.; Volonté, D.; Minetti, C.; Bregman, D.B.; Lisanti, M.P. Limb-girdle muscular dystrophy (LGMD-1C) mutants of caveolin-3 undergo ubiquitination and proteasomal degradation. Treatment with proteasomal inhibitors blocks the dominant negative effect of LGMD-1C mutants and rescues wild-type caveolin-3. J. Biol. Chem. 2000, 275, 37702–37711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, A.; Evesson, F.J.; Sutton, R.B.; North, K.N.; Cooper, S.T. Ferlins: regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair. Traffic 2012, 13, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Therrien, C.; Di Fulvio, S.; Pickles, S.; Sinnreich, M. Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry 2009, 48, 2377–2384. [Google Scholar] [CrossRef]
- Abdullah, N.; Padmanarayana, M.; Marty, N.J.; Johnson, C.P. Quantitation of the calcium and membrane binding properties of the C2 domains of dysferlin. Biophys. J. 2014, 106, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, J.D.; Lim, N.K.; Schenck, S.; Duerst, A.; Dutzler, R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 2014, 516, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Galietta, L.J.V. Structure and function of tmem16 proteins (anoctamins). Physiol. Rev. 2014, 94, 419–459. [Google Scholar] [CrossRef] [Green Version]
- Picollo, A.; Malvezzi, M.; Accardi, A. TMEM16 proteins: Unknown structure and confusing functions. J. Mol. Biol. 2015, 427, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, J.M.; Hartzell, H.C. Anoctamins/TMEM16 proteins: Chloride channels flirting with lipids and extracellular vesicles. Annu. Rev. Physiol. 2017, 79, 119–143. [Google Scholar] [CrossRef] [Green Version]
- Falzone, M.E.; Malvezzi, M.; Lee, B.C.; Accardi, A. Known structures and unknown mechanisms of TMEM16 scramblases and channels. J. Gen. Physiol. 2018, 150, 933–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuta, K.; Tsutsumi, S.; Inoue, H.; Sakamoto, Y.; Miyatake, K.; Miyawaki, K.; Noji, S.; Kamata, N.; Itakura, M. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem. Biophys. Res. Commun. 2007, 357, 126–132. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Inoue, H.; Sakamoto, Y.; Mizuta, K.; Kamata, N.; Itakura, M. Molecular cloning and characterization of the murine gnathodiaphyseal dysplasia gene GDD1. Biochem. Biophys. Res. Commun. 2005, 331, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Gyobu, S.; Miyata, H.; Ikawa, M.; Yamazaki, D.; Takeshima, H.; Suzuki, J.; Nagata, S. A role of TMEM16E carrying a scrambling domain in sperm motility. Mol. Cell. Biol. 2016, 36, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, K.L.B. RING fingers and B-boxes: Zinc-binding protein-protein interaction domains. Biochem. Cell Biol. 1998, 76, 351–358. [Google Scholar] [CrossRef]
- Levy, J.R.; Campbell, K.P.; Glass, D.J. MG53’s new identity. Skelet. Muscle 2013, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Sorimachi, H. Calpains—An elaborate proteolytic system. Biochim. Biophys. Acta Proteins Proteomics 2012, 1824, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Moss, S.E.; Morgan, R.O. The annexins. Genome Biol. 2004, 5, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Swairjo, M.A.; Seaton, B.A. Annexin structure and membrane interactions: A molecular perspective. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 193–213. [Google Scholar] [CrossRef]
- Koerdt, S.N.; Ashraf, A.P.K.; Gerke, V. Annexins and plasma membrane repair. In Current Topics in Membranes; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 84, pp. 43–65. ISBN 9780128177600. [Google Scholar]
- Bendix, P.M.; Simonsen, A.C.; Florentsen, C.D.; Häger, S.C.; Mularski, A.; Zanjani, A.A.H.; Moreno-Pescador, G.; Klenow, M.B.; Sønder, S.L.; Danielsen, H.M.; et al. Interdisciplinary synergy to reveal mechanisms of annexin-mediated plasma membrane shaping and repair. Cells 2020, 9, 1029. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Berendes, R.; Burger, A.; Schneider, M.; Karshikov, A.; Luecke, H.; Römisch, J.; Paques, E. Crystal and molecular structure of human annexin V after refinement. Implications for structure, membrane binding and ion channel formation of the annexin family of proteins. J. Mol. Biol. 1992, 223, 683–704. [Google Scholar] [CrossRef]
- Madej, T.; Lanczycki, C.J.; Zhang, D.; Thiessen, P.A.; Geer, R.C.; Marchler-Bauer, A.; Bryant, S.H. MMDB and VAST+: Tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014, 42, D297–D303. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, R.A.; Ernst, J.D. Characterization of Ca2(+)-dependent phospholipid binding, vesicle aggregation and membrane fusion by annexins. Biochem. J. 1990, 266, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, O.; Gerke, V.; Bader, M.F.; Porte, F.; Brisson, A. Structural analysis of junctions formed between lipid membranes and several annexins by cryo-electron microscopy. J. Mol. Biol. 1997, 272, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Bitto, E.; Cho, W. Structural determinant of the vesicle aggregation activity of annexin I. Biochemistry 1999, 38, 14094–14100. [Google Scholar] [CrossRef]
- Rintala-Dempsey, A.C.; Rezvanpour, A.; Shaw, G.S. S100-annexin complexes—Structural insights. FEBS J. 2008, 275, 4956–4966. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Parra, A.V. Vesicle aggregation by annexin I: Role of a secondary membrane binding site. Biochemistry 1995, 34, 10393–10399. [Google Scholar] [CrossRef]
- Ayala-Sanmartin, J.; Zibouche, M.; Illien, F.; Vincent, M.; Gallay, J. Insight into the location and dynamics of the annexin A2 N-terminal domain during Ca2+-induced membrane bridging. Biochim. Biophys. Acta Biomembr. 2008, 1778, 472–482. [Google Scholar] [CrossRef]
- Lauritzen, S.P.; Boye, T.L.; Nylandsted, J. Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin. Cell Dev. Biol. 2015, 45, 32–38. [Google Scholar] [CrossRef]
- Kaetzel, M.A.; Mo, Y.D.; Mealy, T.R.; Campos, B.; Bergsma-Schutter, W.; Brisson, A.; Dedman, J.R.; Seaton, B.A. Phosphorylation mutants elucidate the mechanism of annexin IV-mediated membrane aggregation. Biochemistry 2001, 40, 4192–4199. [Google Scholar] [CrossRef]
- Crosby, K.C.; Postma, M.; Hink, M.A.; Zeelenberg, C.H.C.; Adjobo-Hermans, M.J.W.; Gadella, T.W.J. Quantitative analysis of self-association and mobility of Annexin A4 at the plasma membrane. Biophys. J. 2013, 104, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govorukhina, N.; Bergsma-schutter, W.; Mazères-dubut, C.; Mazères, S.; Drakopoulou, E.; Bystrykh, L.; Oling, F.; Mukhopadhyay, A.; Reviakine, I.; Lai, J.; et al. Self-assembly of annexin A5 on lipid membranes. In Annexins: Biological Importance and Annexin-Related Pathologies; Landes Biosciences: Georgetown, TX, USA, 2003; pp. 61–66. [Google Scholar]
- Buzhynskyy, N.; Golczak, M.; Lai-Kee-Him, J.; Lambert, O.; Tessier, B.; Gounou, C.; Bérat, R.; Simon, A.; Granier, T.; Chevalier, J.M.; et al. Annexin-A6 presents two modes of association with phospholipid membranes. A combined QCM-D, AFM and cryo-TEM study. J. Struct. Biol. 2009, 168, 107–116. [Google Scholar] [CrossRef]
- Boye, T.L.; Jeppesen, J.C.; Maeda, K.; Pezeshkian, W.; Solovyeva, V.; Nylandsted, J.; Simonsen, A.C. Annexins induce curvature on free-edge membranes displaying distinct morphologies. Sci. Rep. 2018, 8, 10309. [Google Scholar] [CrossRef] [PubMed]
- Creutz, C.E.; Pazoles, C.J.; Pollard, H.B. Identification and purification of an adrenal medullary protein (synexin) that causes calcium-dependent aggregation of isolated chromaffin granules. J. Biol. Chem. 1978, 253, 2858–2866. [Google Scholar] [CrossRef]
- Selbert, S.; Fischer, P.; Pongratz, D.; Stewart, M.; Noegel, A.A. Expression and localization of annexin VII (synexin) in muscle cells. J. Cell Sci. 1995, 108, 85–95. [Google Scholar] [CrossRef]
- Pollard, H.B.; Lee Burns, A.; Rojas, E. Synexin (Annexin VII): A cytosolic calcium-binding protein which promotes membrane fusion and forms calcium channels in artificial bilayer and natural membranes. J. Membr. Biol. 1990, 117, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Beckmann, J. 30th and 31st ENMC international workshops, Naarden, The Netherlands, Held 6–8 January 1995. Neuromuscul. Disord. 1995, 5, 337–343. [Google Scholar] [CrossRef]
- Wenzel, K.; Geier, C.; Qadri, F.; Hubner, N.; Schulz, H.; Erdmann, B.; Gross, V.; Bauer, D.; Dechend, R.; Dietz, R.; et al. Dysfunction of dysferlin-deficient hearts. J. Mol. Med. 2007, 85, 1203–1214. [Google Scholar] [CrossRef]
- Schilling, J.M.; Patel, H.H. Non-canonical roles for caveolin in regulation of membrane repair and mitochondria: Implications for stress adaptation with age. J. Physiol. 2016, 594, 4581–4589. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.C.; Braun, C.; Vorgerd, M.; Poppe, M.; Thirion, C.; Schmidt, C.; Schreiber, H.; Knirsch, U.I.; Brummer, D.; Müller-Felber, W.; et al. Variable reduction of caveolin-3 in patients with LGMD2B/MM. J. Neurol. 2003, 250, 1431–1438. [Google Scholar] [CrossRef]
- Mcneil, P.L.; Terasaki, M. Coping with the inevitable: How cells repair a torn surface membrane. Nat. Cell Biol. 2001, 3, E124–E129. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. Muscular dystrophy. Curr. Opin. Pediatr. 2013, 25, 701–707. [Google Scholar] [CrossRef]
- Vilchez, J.J.; Gallano, P.; Gallardo, E.; Lasa, A.; Rojas-García, R.; Freixas, A.; De Luna, N.; Calafell, F.; Sevilla, T.; Mayordomo, F.; et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the spanish population. Arch. Neurol. 2005, 62, 1256–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydemann, A.; Huber, J.M.; Demonbreun, A.; Hadhazy, M.; McNally, E.M. Genetic background influences muscular dystrophy. Neuromuscul. Disord. 2005, 15, 601–609. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Capote, J.; Ohiri, J.C.; Warner, J.L.; Vo, A.H.; Earley, J.U.; Hadhazy, M.; Demonbreun, A.R.; Spencer, M.J.; McNally, E.M. Genetic modifiers of muscular dystrophy act on sarcolemmal resealing and recovery from injury. PLoS Genet. 2017, 13, e1007070. [Google Scholar] [CrossRef]
- Cagliani, R.; Magri, F.; Toscano, A.; Merlini, L.; Fortunato, F.; Lamperti, C.; Rodolico, C.; Prelle, A.; Sironi, M.; Aguennouz, M.; et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum. Mutat. 2005, 26, 283. [Google Scholar] [CrossRef] [PubMed]
- Kesari, A.; Fukuda, M.; Knoblach, S.; Bashir, R.; Nader, G.A.; Rao, D.; Nagaraju, K.; Hoffman, E.P. Dysferlin deficiency shows compensatory induction of Rab27A/Slp2a that may contribute to inflammatory onset. Am. J. Pathol. 2008, 173, 1476–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, H.T.; Mossakowski, A.A.; Willis, B.J.; Grimsrud, K.N.; Wood, J.A.; Lloyd, K.C.K.; Zbinden-Foncea, H.; Baar, K. Generation of desminopathy in rats using CRISPR-Cas9. J. Cachexia. Sarcopenia Muscle 2020, 11, 1364–1376. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Burkhard, F.C.; Wray, S.; Draeger, A. Modulating signaling events in smooth muscle: Cleavage of annexin 2 abolishes its binding to lipid rafts. FASEB J. 2002, 16, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Mellgren, R.L.; Huang, X. Fetuin A stabilizes m-calpain and facilitates plasma membrane repair. J. Biol. Chem. 2007, 282, 35868–35877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley-Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Allen, M.V.; Warner, J.L.; Barefield, D.Y.; Krishnan, S.; Swanson, K.E.; Earley, J.U.; McNally, E.M. Enhanced muscular dystrophy from loss of dysferlin is accompanied by impaired annexin A6 translocation after sarcolemmal disruption. Am. J. Pathol. 2016, 186, 1610–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Symbol | Function | Binding Partners | Ref |
|---|---|---|---|---|
| Annexin A1 | ANXA1 | The “lipid patch” formation | DYSF, S100A11 | [40,41,42,43] |
| Annexin A2 | ANXA2 | The “lipid patch” formation | DYSF, S100A10 | [40,41,42,43] |
| Annexin A4 | ANXA4 | To be established | To be identified | [44] |
| Annexin A5 | ANXA5 | Strengthening the damaged membrane | To be identified | [24,45] |
| Annexin A6 | ANXA6 | Condensing membranes at the disruption site | To be identified | [26,29,46,47] |
| Annexin A7 | ANXA7 | To be established | To be identified | [48,49] |
| Anoctamin-5 | ANO5 | To be established | To be identified | [50,51,52,53,54] |
| Calpains | CAPN1-3 | The formation of mini-dysferlinc72 | DYSF | [55,56,57,58] |
| Caveolin-3 | CAV3 | Controlling membrane tension | DYSF, MG53 | [59,60,61,62,63,64,65] |
| Dysferlin | DYSF | Recruiting the “lipid patch” | ANXA1, ANXA2, Calpains, CAV3, MG53 | [8,9,40,46,55,59,66,67,68,69,70] |
| Mitsugumin-53 | MG53 | Strengthening the damaged membrane and recruiting the “lipid patch” | DYSF, CAV3 | [65,70,71] |
| New Nomenclature | Former Nomenclature | MutatedGene | Protein | Ref |
|---|---|---|---|---|
| LGMD D4 Calpain3-related | LGMD1I | CAPN3 | Calpain-3 | [72,73] |
| LGMD R1 Calpain3-related | LGMD2A | CAPN3 | Calpain-3 | [72,73] |
| LGMD R2 Dysferlin-related | LGMD2B | DYSF | Dysferlin | [8,9,66,74,75] |
| LGMD R12 Anoctamin5-related | LGMD2L | ANO5 | Anoctamin-5 | [12,51,76] |
| Rippling muscle disease Caveolin3-related | LGMD1C | CAV3 | Caveolin-3 | [11,77,78,79,80,81] |
| MMD1 or Miyoshi myopathy | MMD1 or Miyoshi myopathy | DYSF | Dysferlin | [8,9,66,74,75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croissant, C.; Carmeille, R.; Brévart, C.; Bouter, A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. Int. J. Mol. Sci. 2021, 22, 5276. https://doi.org/10.3390/ijms22105276
Croissant C, Carmeille R, Brévart C, Bouter A. Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. International Journal of Molecular Sciences. 2021; 22(10):5276. https://doi.org/10.3390/ijms22105276
Chicago/Turabian StyleCroissant, Coralie, Romain Carmeille, Charlotte Brévart, and Anthony Bouter. 2021. "Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies" International Journal of Molecular Sciences 22, no. 10: 5276. https://doi.org/10.3390/ijms22105276
APA StyleCroissant, C., Carmeille, R., Brévart, C., & Bouter, A. (2021). Annexins and Membrane Repair Dysfunctions in Muscular Dystrophies. International Journal of Molecular Sciences, 22(10), 5276. https://doi.org/10.3390/ijms22105276