
Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics




Abstract
:1. Introduction
2. Results
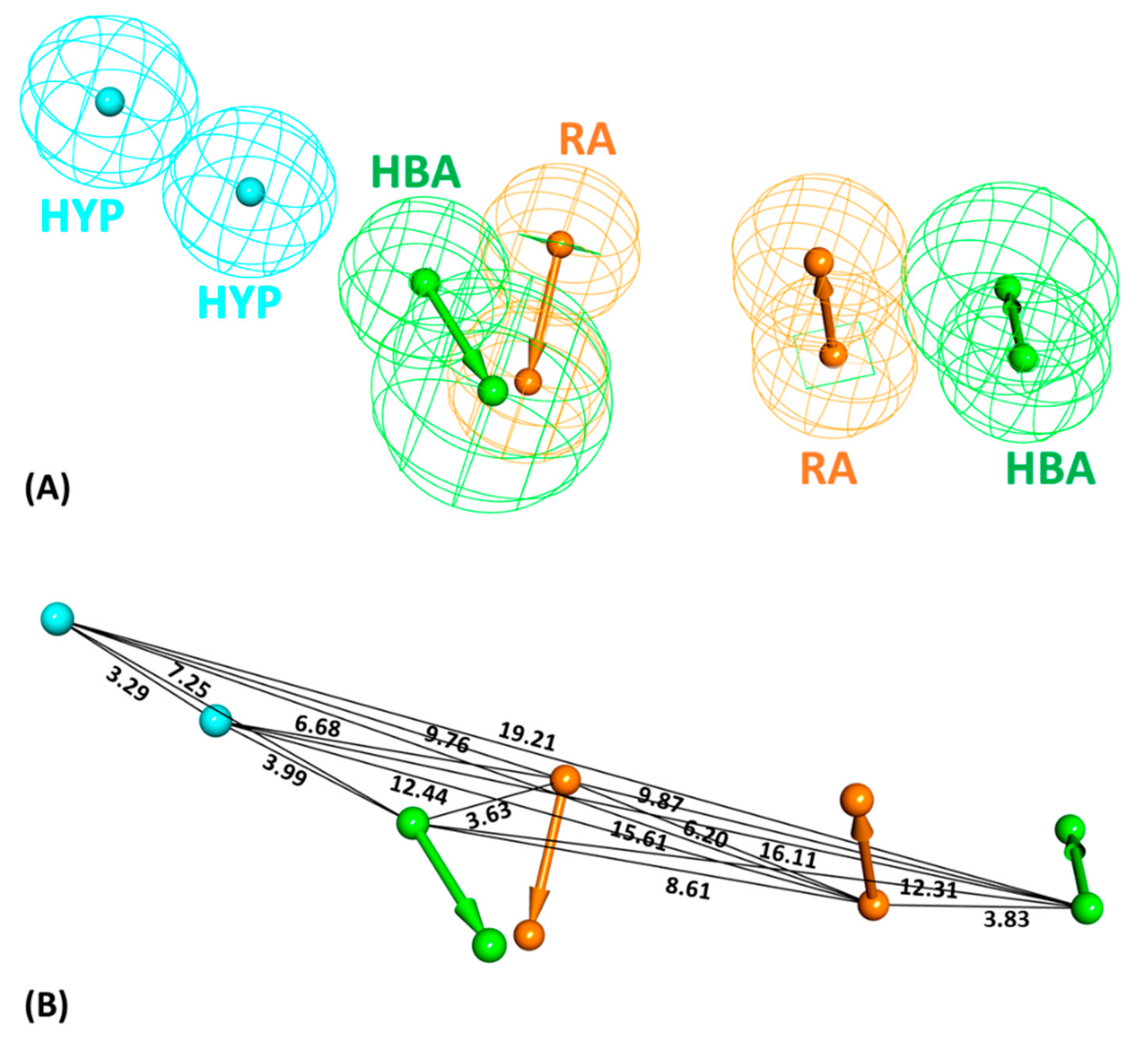
2.1. Common Feature Pharmacophore Model
2.2. Decoy Set Validation of the Pharmacophore Model
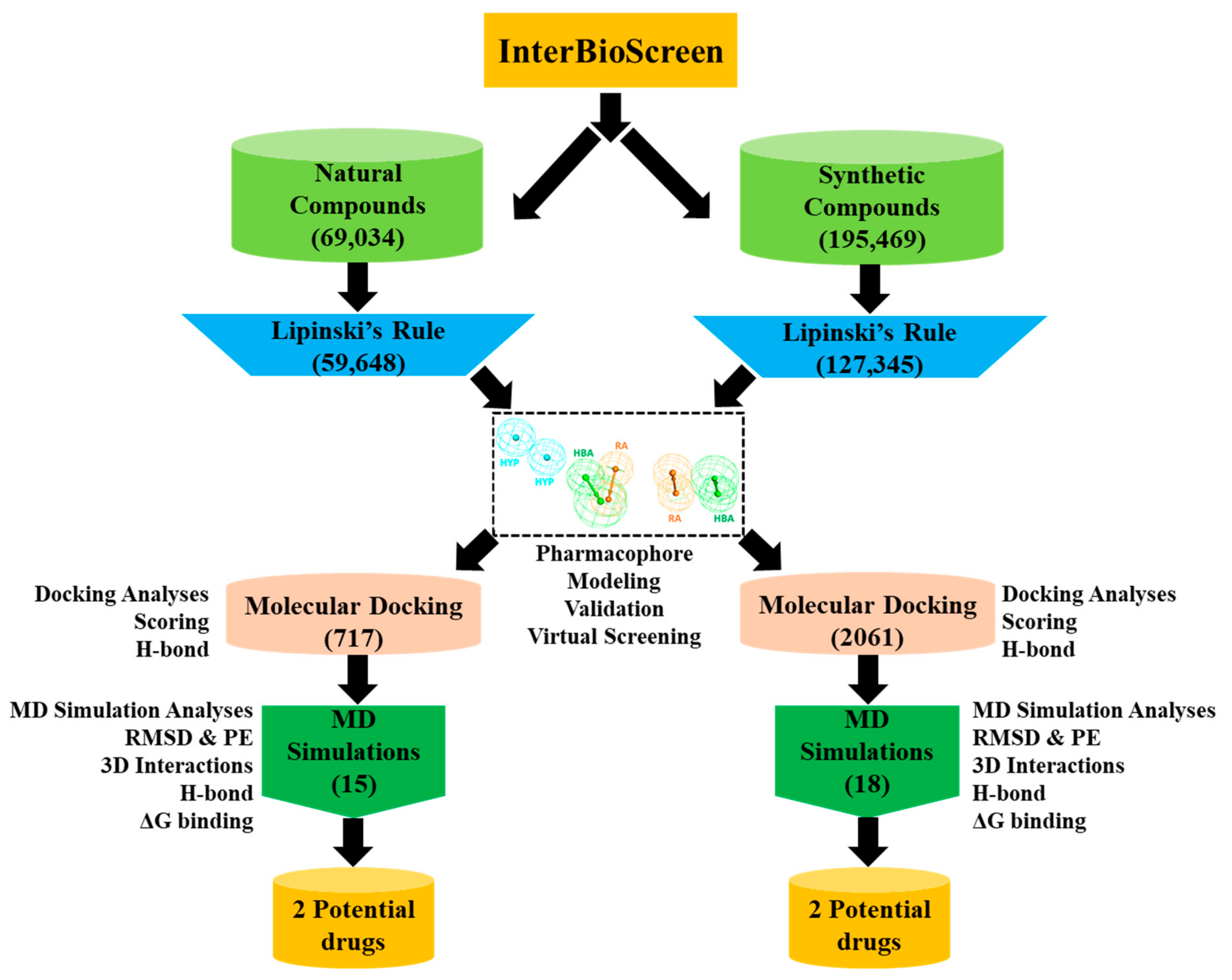
2.3. Drug-Likeness Evaluation and Virtual Screening of InterBioScreen Database
2.4. Molecular Docking of Drug-Like Compounds with Heparanase
2.5. Molecular Dynamics Simulation Analysis
2.5.1. Analysis of Stability and Binding Free Energy
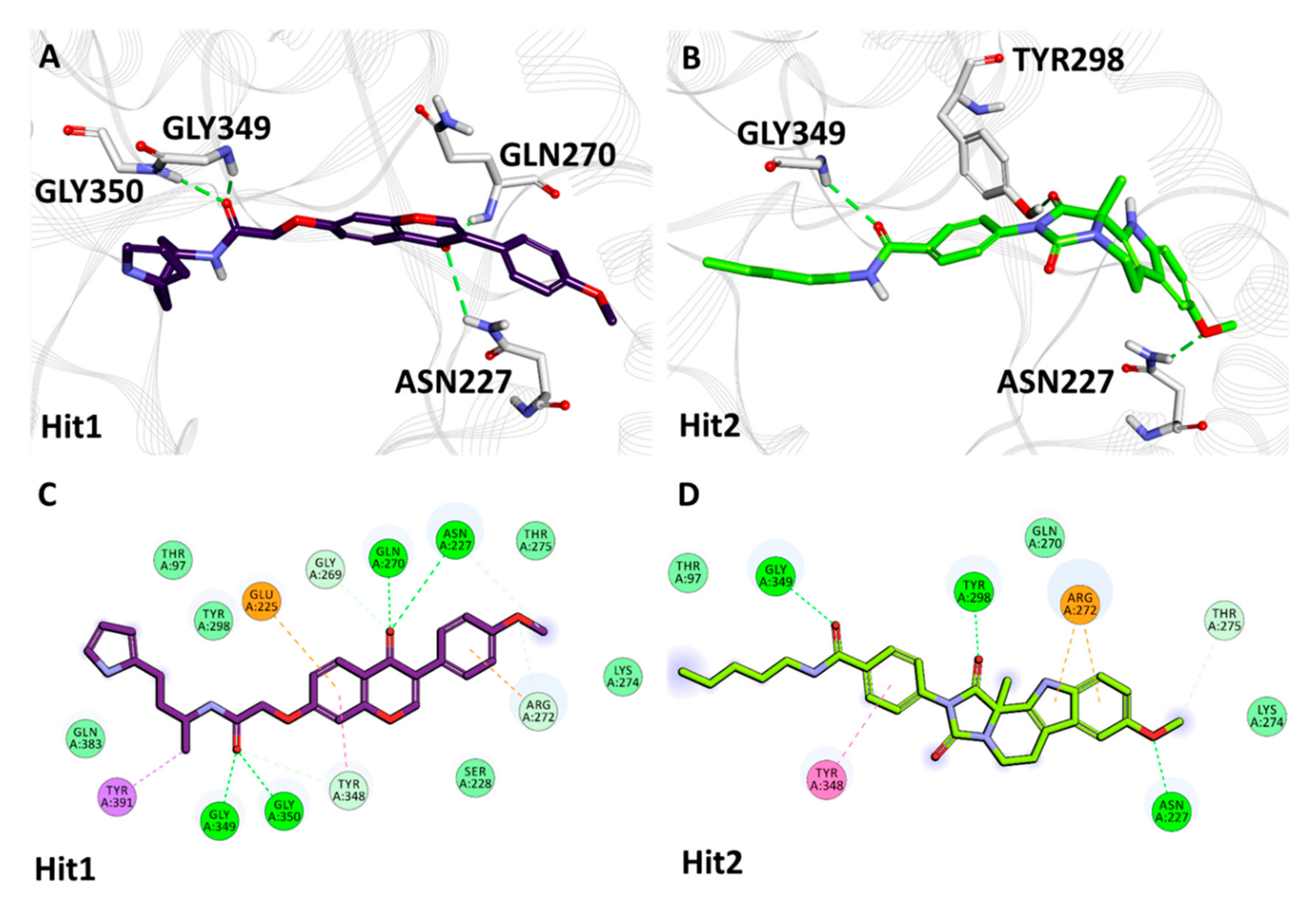
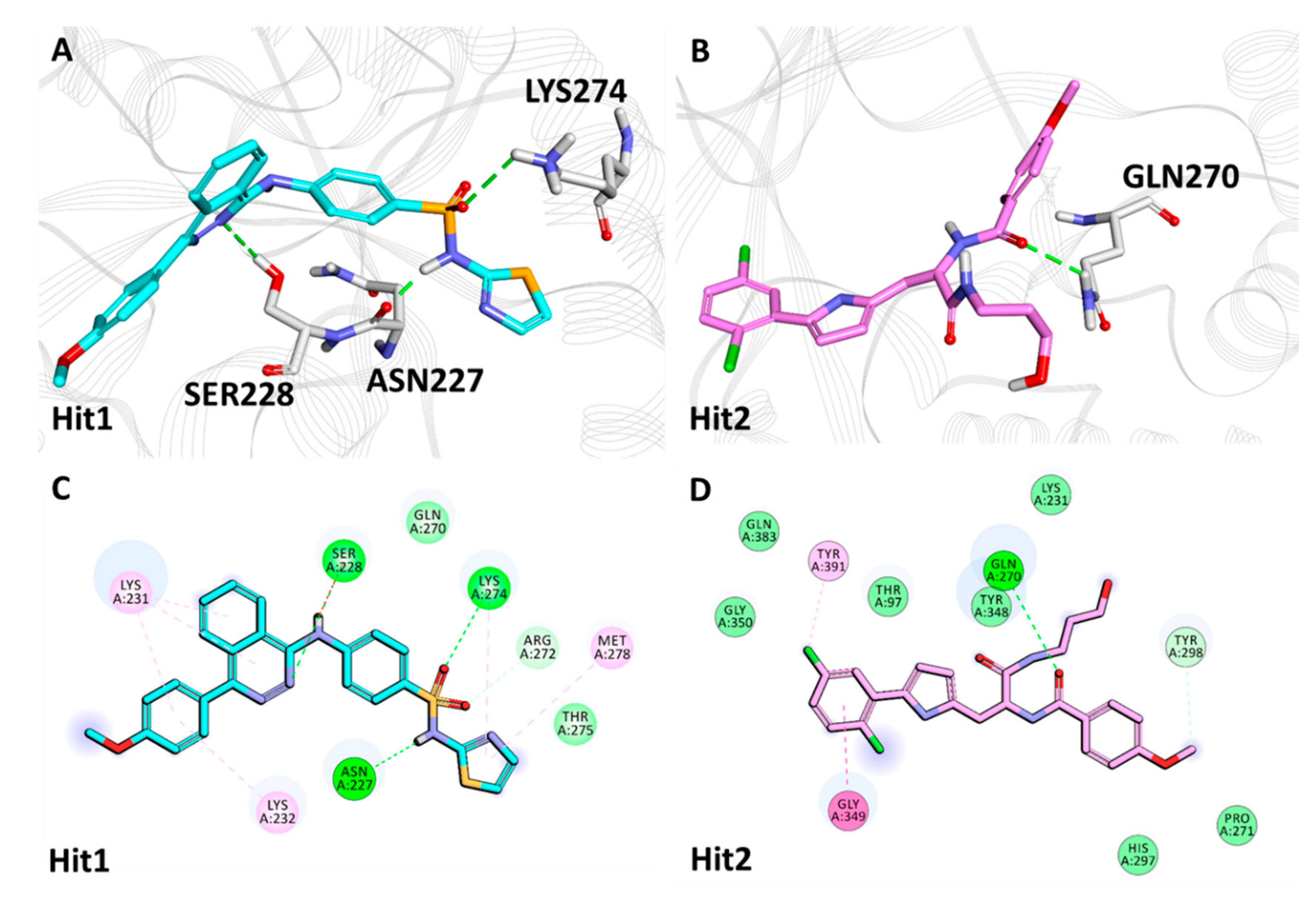
2.5.2. Binding Mode and Molecular Interactions with Heparanase Active Site
3. Discussion
4. Materials and Methods
4.1. Dataset Preparation and Pharmacophore Model Generation
4.2. Validation of the Generated Model
4.3. Drug-Like Database Generation and Virtual Screening of InterBioScreen Database
4.4. Molecular Docking of Screened Drug-Like Compounds with Hpse
4.5. Molecular Dynamics Simulation of Identified Natural and Synthetic Compounds
4.6. Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Basappa; Rangappa, K.S. Identification of Novel Class of Triazolo-Thiadiazoles as Potent Inhibitors of Human Heparanase and their Anticancer Activity. BMC Cancer 2017, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updat. 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Coombe, D.R.; Gandhi, N.S. Heparanase: A Challenging Cancer Drug Target. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Ramani, V.C.; Yang, Y.; Ren, Y.; Nan, L.; Sanderson, R.D. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J. Biol. Chem. 2011, 286, 6490–6499. [Google Scholar] [CrossRef] [Green Version]
- Purushothaman, A.; Chen, L.; Yang, Y.; Sanderson, R.D. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J. Biol. Chem. 2008, 283, 32628–32636. [Google Scholar] [CrossRef] [Green Version]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol. 2006, 38, 2018–2039. [Google Scholar] [CrossRef]
- Cassinelli, G.; Favini, E.; Bo, L.D.; Tortoreto, M.; De Maglie, M.; Dagrada, G.; Pilotti, S.; Zunino, F.; Zaffaroni, N.; Lanzi, C. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget 2016, 7, 47848–47863. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012, 5, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Pisano, C.; Vlodavsky, I.; Ilan, N.; Zunino, F. The potential of heparanase as a therapeutic target in cancer. Biochem. Pharmacol. 2014, 89, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef] [Green Version]
- Khasraw, M.; Pavlakis, N.; McCowatt, S.; Underhill, C.; Begbie, S.; de Souza, P.; Boyce, A.; Parnis, F.; Lim, V.; Harvie, R.; et al. Multicentre phase I/II study of PI-88, a heparanase inhibitor in combination with docetaxel in patients with metastatic castrate-resistant prostate cancer. Ann. Oncol. 2009, 21, 1302–1307. [Google Scholar] [CrossRef]
- A Phase III PI-88 in the Adjuvant Treatment of Subjects with Hepatitis Virus Related HCC After Surgical Resection—Tabular View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01402908 (accessed on 27 February 2021).
- SST0001 (Roneparstat) in Advanced Multiple Myeloma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01764880 (accessed on 27 February 2021).
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Study of the Safety and Tolerability of IV Infused PG545 in Patients with Advanced Solid Tumours—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02042781 (accessed on 27 February 2021).
- Dredge, K.; Hammond, E.; Handley, P.; Gonda, T.J.; Smith, M.T.; Vincent, C.; Brandt, R.; Ferro, V.; Bytheway, I. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br. J. Cancer 2011, 104, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, a novel Heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef] [Green Version]
- M402 in Combination with Nab-Paclitaxel and Gemcitabine in Pancreatic Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01621243 (accessed on 27 February 2021).
- Zhao, H.; Liu, H.; Chen, Y.; Xin, X.; Li, J.; Hou, Y.; Zhang, Z.; Zhang, X.; Xie, C.; Geng, M.; et al. Oligomannurarate sulfate, a novel heparanase inhibitor simultaneously targeting basic fibroblast growth factor, combats tumor angiogenesis and metastasis. Cancer Res. 2006, 66, 8779–8787. [Google Scholar] [CrossRef] [Green Version]
- Parish, C.R.; Coombe, D.R.; Jakobsen, K.B.; Bennett, F.A.; Underwood, P.A. Evidence that sulphated polysaccharides inhibit tumour metastasis by blocking tumour-cell-derived heparanases. Int. J. Cancer 1987, 40, 511–518. [Google Scholar] [CrossRef]
- Ishida, K.; Hirai, G.; Murakami, K.; Teruya, T.; Simizu, S.; Sodeoka, M.; Osada, H. Structure-based design of a selective heparanase inhibitor as an antimetastatic agent. Mol. Cancer Ther. 2004, 3, 1069–1077. [Google Scholar]
- Hamaguchi, T.; Sudo, T.; Osada, H. RK-682, a potent inhibitor of tyrosine phosphatase, arrested the mammalian cell cycle progression at G1phase. FEBS Lett. 1995, 372, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Heyman, B.; Yang, Y. Mechanisms of heparanase inhibitors in cancer therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Echart, C.; Distaso, M.; Baraz, L.; Vlodavsky, I.; Iacobelli, M. Defibrotide downregulates Heparanase expression in multiple myeloma, renal adenocarcinoma and breast cancer cell lines. Cancer Res. 2008, 68, 1180. [Google Scholar]
- Madia, V.N.; Messore, A.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; Scipione, L.; Costi, R.; Rivara, S.; et al. Novel Benzazole Derivatives Endowed with Potent Antiheparanase Activity. J. Med. Chem. 2018, 61, 6918–6936. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Suramin: A potent inhibitor of the reverse transcriptase of RNA tumor viruses. Cancer Lett. 1979, 8, 9–22. [Google Scholar] [CrossRef]
- Nakajima, M.; DeChavigny, A.; Johnson, C.E.; Hamada, J.I.; Stein, C.A.; Nicolson, G.L. Suramin: A potent inhibitor of melanoma heparanase and invasion. J. Biol. Chem. 1991, 266, 9661–9666. [Google Scholar] [CrossRef]
- Pan, W.; Miao, H.Q.; Xu, Y.J.; Navarro, E.C.; Tonra, J.R.; Corcoran, E.; Lahiji, A.; Kussie, P.; Kiselyov, A.S.; Wong, W.C.; et al. 1-[4-(1H-Benzoimidazol-2-yl)-phenyl]-3-[4-(1H-benzoimidazol-2-yl)-phenyl] -urea derivatives as small molecule heparanase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Miao, H.Q.; Pan, W.; Navarro, E.C.; Tonra, J.R.; Mitelman, S.; Camara, M.M.; Deevi, D.S.; Kiselyov, A.S.; Kussie, P.; et al. N-(4-{[4-(1H-Benzoimidazol-2-yl)-arylamino]-methyl}-phenyl)-benzamide derivatives as small molecule heparanase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 404–408. [Google Scholar] [CrossRef]
- Courtney, S.M.; Hay, P.A.; Buck, R.T.; Colville, C.S.; Porter, D.W.; Scopes, D.I.C.; Pollard, F.C.; Page, M.J.; Bennett, J.M.; Hircock, M.L.; et al. 2,3-Dihydro-1,3-dioxo-1H-isoindole-5-carboxylic acid derivatives: A novel class of small molecule heparanase inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 3269–3273. [Google Scholar] [CrossRef]
- Courtney, S.M.; Hay, P.A.; Buck, R.T.; Colville, C.S.; Phillips, D.J.; Scopes, D.I.C.; Pollard, F.C.; Page, M.J.; Bennett, J.M.; Hircock, M.L.; et al. Furanyl-1,3-thiazol-2-yl and benzoxazol-5-yl acetic acid derivatives: Novel classes of heparanase inhibitor. Bioorganic Med. Chem. Lett. 2005, 15, 2295–2299. [Google Scholar] [CrossRef]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus Kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [Green Version]
- Bharathkumar, H.; Mohan, C.D.; Rangappa, S.; Kang, T.; Keerthy, H.K.; Fuchs, J.E.; Kwon, N.H.; Bender, A.; Kim, S.; Basappa; et al. Screening of quinoline, 1,3-benzoxazine, and 1,3-oxazine-based small molecules against isolated methionyl-tRNA synthetase and A549 and HCT116 cancer cells including an in silico binding mode analysis. Org. Biomol. Chem. 2015, 13, 9381–9387. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, V.; Mohan, C.D.; Baburajeev, C.P.; Rangappa, S.; Jagadish, S.; Fuchs, J.E.; Sukhorukov, A.Y.; Chandra; Mason, D.J.; Sharath-Kumar, K.S.; et al. Synthesis and characterization of novel oxazines and demonstration that they specifically target cyclooxygenase 2. Bioorg. Med. Chem. Lett. 2015, 25, 2931–2936. [Google Scholar] [CrossRef]
- Nirvanappa, A.C.; Mohan, C.D.; Rangappa, S.; Ananda, H.; Sukhorukov, A.Y.; Shanmugam, M.K.; Sundaram, M.S.; Nayaka, S.C.; Girish, K.S.; Chinnathambi, A.; et al. Novel Synthetic Oxazines Target NF-κB in Colon Cancer In Vitro and Inflammatory Bowel Disease In Vivo. PLoS ONE 2016, 11, e0163209. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, N.B.; Mohan, C.D.; Basappa, B.; Pandey, V.; Rangappa, S.; Bharathkumar, H.; Kumar, A.P.; Lobie, P.E.; Rangappa, K.S. An azaspirane derivative suppresses growth and induces apoptosis of ER-positive and ER-negative breast cancer cells through the modulation of JAK2/STAT3 signaling pathway. Int. J. Oncol. 2016, 49, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Basappa; Murugan, S.; Kavitha, C.V.; Purushothaman, A.; Nevin, K.G.; Sugahara, K.; Rangappa, K.S. A small oxazine compound as an anti-tumor agent: A novel pyranoside mimetic that binds to VEGF, HB-EGF, and TNF-α. Cancer Lett. 2010, 297, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Uzma, F.; Mohan, C.D.; Hashem, A.; Konappa, N.M.; Rangappa, S.; Kamath, P.V.; Singh, B.P.; Mudili, V.; Gupta, V.K.; Siddaiah, C.N.; et al. Endophytic fungi-alternative sources of cytotoxic compounds: A review. Front. Pharmacol. 2018, 9, 309. [Google Scholar] [CrossRef]
- Gozalbes, R.; Mosulén, S.; Ortí, L.; Rodríguez-Díaz, J.; Carbajo, R.J.; Melnyk, P.; Pineda-Lucena, A. Hit identification of novel heparanase inhibitors by structure- and ligand-based approaches. Bioorg. Med. Chem. 2013, 21, 1944–1951. [Google Scholar] [CrossRef]
- Gozalbes, R.; Mosulén, S.; Carbajo, R.J.; Pineda-Lucena, A. Development and NMR validation of minimal pharmacophore hypotheses for the generation of fragment libraries enriched in heparanase inhibitors. J. Comput. Mol. Des. 2009, 23, 555–569. [Google Scholar] [CrossRef]
- Dai, X.; Yan, J.; Fu, X.; Pan, Q.; Sun, D.; Xu, Y.; Wang, J.; Nie, L.; Tong, L.; Shen, A.; et al. Aspirin inhibits cancer metastasis and angiogenesis via targeting heparanase. Clin. Cancer Res. 2017, 23, 6267–6279. [Google Scholar] [CrossRef] [Green Version]
- Parate, S.; Kumar, V.; Hong, J.C.; Lee, K.W. Identification of Flavonoids as Putative ROS-1 Kinase Inhibitors Using Pharmacophore Modeling for NSCLC Therapeutics. Molecules 2021, 26, 2114. [Google Scholar] [CrossRef]
- Parate, S.; Kumar, V.; Lee, G.; Rampogu, S.; Hong, J.C.; Lee, K.W. Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals 2021, 14, 282. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Jayatilleke, K.M.; Hulett, M.D. Heparanase and the hallmarks of cancer. J. Transl. Med. 2020, 18, 1–25. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Ilan, N.; Naggi, A.; Casu, B. Heparanase: Structure, Biological Functions, and Inhibition by Heparin-Derived Mimetics of Heparan Sulfate. Curr. Pharm. Des. 2007, 13, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, Y.; Guner, O. Pharmacophore Modeling and Three-dimensional Database Searching for Drug Design Using Catalyst. Curr. Med. Chem. 2012, 8, 1035–1055. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.; Gillet, V.J.; Bravi, G.; Leach, A.R. A comparison of the pharmacophore identification programs: Catalyst, DISCO and GASP. J. Comput. Mol. Des. 2002, 16, 653–681. [Google Scholar] [CrossRef] [Green Version]
- Ataei, S.; Yilmaz, S.; Ertan-Bolelli, T.; Yildiz, I. Generated 3D-Common Feature Hypotheses Using the HipHop Method for Developing New Topoisomerase i Inhibitors. Arch. Pharm. 2015, 348, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Purushottamachar, P.; Patel, J.B.; Gediya, L.K.; Clement, O.O.; Njar, V.C.O. First chemical feature-based pharmacophore modeling of potent retinoidal retinoic acid metabolism blocking agents (RAMBAs): Identification of novel RAMBA scaffolds. Eur. J. Med. Chem. 2012, 47, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Kumar, R.; Parate, S.; Yoon, S.; Lee, G.; Kim, D.; Lee, K.W. Identification of ACK1 Inhibitors as Anticancer Agents by using Computer-Aided Drug Designing. J. Mol. Struct. 2021, 1235, 1. [Google Scholar] [CrossRef]
- Zeb, A.; Kim, D.; Alam, S.I.; Son, M.; Kumar, R.; Rampogu, S.; Parameswaran, S.; Shelake, R.M.; Rana, R.M.; Parate, S.; et al. Computational Simulations Identify Pyrrolidine-2,3-Dione Derivatives as Novel Inhibitors of Cdk5/p25 Complex to Attenuate Alzheimer’s Pathology. J. Clin. Med. 2019, 8, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampogu, S.; Parate, S.; Parameswaran, S.; Park, C.; Baek, A.; Son, M.; Park, Y.; Park, S.J.; Lee, K.W. Natural compounds as potential Hsp90 inhibitors for breast cancer-Pharmacophore guided molecular modelling studies. Comput. Biol. Chem. 2019, 83, 107113. [Google Scholar] [CrossRef]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [Green Version]
- Messore, A.; Madia, V.N.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; De Vita, D.; Scipione, L.; Pepi, F.; et al. Novel Symmetrical Benzazolyl Derivatives Endowed with Potent Anti-Heparanase Activity. J. Med. Chem. 2018, 61, 10834–10859. [Google Scholar] [CrossRef] [Green Version]
- Pala, D.; Scalvini, L.; Elisi, G.M.; Lodola, A.; Mor, M.; Spadoni, G.; Ferrara, F.F.; Pavoni, E.; Roscilli, G.; Milazzo, F.M.; et al. New classes of potent heparanase inhibitors from ligand-based virtual screening. J. Enzym. Inhib. Med. Chem. 2020, 35, 1685–1696. [Google Scholar] [CrossRef]
- Pala, D.; Rivara, S.; Mor, M.; Milazzo, F.M.; Roscilli, G.; Pavoni, E.; Giannini, G. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology 2016, 26, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Awortwe, C.; Fasinu, P.S.; Rosenkranz, B. Application of Caco-2 cell line in herb-drug interaction studies: Current approaches and challenges. J. Pharm. Pharm. Sci. 2014, 17, 1–19. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Jiao, Z.; Hu, P.; Xu, H.; Wang, Q. Machine learning and deep learning in chemical health and safety: A systematic review of techniques and applications. J. Chem. Heal. Saf. 2020, 27, 316–334. [Google Scholar] [CrossRef]
- Cadow, J.; Born, J.; Manica, M.; Oskooei, A.; Martínez, M.R. PaccMann: A web service for interpretable anticancer compound sensitivity prediction. Nucleic Acids Res. 2021, 48, W502–W508. [Google Scholar] [CrossRef]
- Thirunavukkarasu, M.K.; Shin, W.H.; Karuppasamy, R. Exploring safe and potent bioactives for the treatment of non-small cell lung cancer. 3 Biotech. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-K. Pharmacophore Perception, Development and Use in Drug Design. Edited by Osman F. Güner. Molecules 2000, 5, 987–989. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Parate, S.; Yoon, S.; Lee, G.; Lee, K.W. Computational Simulations Identified Marine-Derived Natural Bioactive Compounds as Replication Inhibitors of SARS-CoV-2. Front. Microbiol. 2021, 12, 583. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, F.R. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Kumar, V.; Lee, K.W. A computational drug repurposing approach in identifying the cephalosporin antibiotic and anti-hepatitis C drug derivatives for COVID-19 treatment. Comput. Biol. Med. 2021, 130, 104186. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lopes, P.E.M.; MacKerell, A.D. Recent developments and applications of the CHARMM force fields. WIREs Comput. Mol. Sci. 2012, 2, 167–185. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Jaidhan, B.J.; Rao, P.S.; Apparao, A. Energy Minimization and Conformation Analysis of Molecules using Steepest Descent Method. Int. J. Comput. Sci. Inf. Technol. 2014, 5, 3525–3538. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rifai, E.A.; Van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1-Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Features a | Rank b | Direct Hit c | Partial Hit d | Max Fit e |
|---|---|---|---|---|---|
| 1 | RA, RA, HBA, HBA, HYP, HYP | 71.08 | 1111 | 0000 | 6 |
| 2 | RA, RA, HBA, HBA, HYP, HYP | 70.28 | 1111 | 0000 | 6 |
| 3 | RA, RA, HBA, HBA, HYP, HYP | 70.28 | 1111 | 0000 | 6 |
| 4 | RA, RA, HBA, HBA, HYP, HYP | 69.48 | 1111 | 0000 | 6 |
| 5 | RA, HBA, HYP, HYP, HYPA | 66.18 | 1111 | 0000 | 5 |
| 6 | HBA, HYP, HYP, HYPA, HYPA | 66.09 | 1111 | 0000 | 5 |
| 7 | RA, HBA, HYP, HYP, HYPA | 66.04 | 1111 | 0000 | 5 |
| 8 | RA, HBA, HYP, HYP, HYPA | 66.00 | 1111 | 0000 | 5 |
| 9 | RA, RA, HBA, HYP, HYP | 65.96 | 1111 | 0000 | 5 |
| 10 | RA, RA, HBA, HYP, HYP | 65.96 | 1111 | 0000 | 5 |
| Sr. No. | Parameters | Values |
|---|---|---|
| 1 | Total number of compounds in the database (D) | 100 |
| 2 | Total number of active compounds in the database (A) | 4 |
| 3 | Total number of hits retrieved by pharmacophore model from the database (Ht) | 6 |
| 4 | Total number of active compounds in the hit list (Ha) | 4 |
| 5 | % Yield of active ((Ha/Ht) × 100) | 66.66% |
| 6 | % Ratio of actives ((Ha/A) × 100) | 100% |
| 7 | False negatives (A-Ha) | 0 |
| 8 | False positives (Ht-Ha) | 2 |
| 9 | Goodness of fit score (GF) | 0.72 |
| Ligands (IBS ID/REF No.) | Docking Scores | MD Analyses | |||
|---|---|---|---|---|---|
| Goldscore | Chemscore | RMSD (Backbone) | Hydrogen Bond (Å) | Binding Free Energy (kJ/mol) | |
| Natural Compound Hits | |||||
| Hit1 (STOCK1N-70463) | 68.95 | −32.00 | 0.16 | 2.16 | −104.579 ± 20.649 |
| Hit2 (STOCK1N-48729) | 67.79 | −30.66 | 0.15 | 0.98 | −83.751 ± 26.469 |
| Synthetic Compound Hits | |||||
| Hit1 (STOCK1S-95244) | 74.92 | −30.70 | 0.16 | 0.37 | −96.193 ± 23.866 |
| Hit2 (STOCK1S-71515) | 67.53 | −33.38 | 0.14 | 1.17 | −86.806 ± 26.536 |
| Reference Inhibitors | |||||
| REF1 | 55.30 | −24.35 | 0.14 | 0.25 | −74.612 ± 20.900 |
| REF2 | 67.43 | −24.35 | 0.15 | 1.13 | −83.519 ± 31.504 |
| Complex Name | Hydrogen Bond Interactions | van der Waals Interactions | π-π/π-alkyl Interactions | ||||
|---|---|---|---|---|---|---|---|
| Amino Acid | Amino Acid Atom | Ligand Atom | Distance (<3.05 Å) | ||||
| Natural Compound Hits | |||||||
| Heparanase + Natural Compounds | Hit1 | Asn227 | HD22 | O13 | 3.02 | Thr97, Ser228, Gly269, Arg272, Lys274, Thr275, Tyr298, Tyr348, Gln383 | Glu225, Tyr391 |
| Gln270 | HN | O13 | 2.08 | ||||
| Gly349 | HN | O18 | 1.90 | ||||
| Gly350 | HN | O18 | 2.29 | ||||
| Hit2 | Asn227 | HD21 | O29 | 2.07 | Thr97, Gln270, Lys274, Thr275, Gly350 | Arg272, Tyr348 | |
| Tyr298 | HH | O16 | 1.80 | ||||
| Gly349 | HN | O19 | 2.61 | ||||
| Synthetic Compound Hits | |||||||
| Heparanase + Synthetic Compounds | Hit1 | Asn227 | O | H35 | 1.85 | Gln270, Arg272, Thr275 | Lys231, Lys232, Met278 |
| Ser228 | HG | N6 | 2.68 | ||||
| Lys274 | HZ2 | O15 | 2.68 | ||||
| Hit2 | Gln270 | HE21 | O15 | 2.60 | Thr97, Lys231, Pro271, His297, Tyr298, Tyr348, Gly350, Gln383 | Gly349, Tyr391 | |
| Reference (REF) Inhibitors | |||||||
| Heparanase + Reference Inhibitors | REF1 | Glu225 | OE2 | H66 | 1.74 | Thr60, Asp62, Gly95, Gly96, Thr97, Ser228, Arg272, His296, Glu343, Gln383, Ala388 | Tyr298, Val384, Tyr391 |
| REF2 | Gln349 | HN | O23 | 2.39 | Thr97, Gln270, Pro271, Tyr348, Gly350, Gln383, Gly389, Asn390 | Arg272, Tyr391 | |
| Hits (IBS a ID) | Mouse Female Carcinogenicity | Mouse Male Carcinogenicity | AMES b Mutagenicity | Skin Irritancy |
|---|---|---|---|---|
| Natural Compound Hits | ||||
| Hit1 (STOCK1N-70463) | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | Non-Irritant |
| Hit2 (STOCK1N-48729) | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | Non-Irritant |
| Synthetic Compound Hits | ||||
| Hit1 (STOCK1S-95244) | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | Non-Irritant |
| Hit2 (STOCK1S-71515) | Non-Carcinogen | Non-Carcinogen | Non-Mutagen | Non-Irritant |
| PK Properties | Natural Compound Hits | Synthetic Compound Hits | Reference Inhibitors | Cut-Off | |||
|---|---|---|---|---|---|---|---|
| Hit1 (STOCK1N-70463) | Hit2 (STOCK1N-48729) | Hit1 (STOCK1S-95244) | Hit2 (STOCK1S-71515) | REF1 | REF2 | ||
| Molecular weight | 447.48 | 474.56 | 489.58 | 489.35 | 500.60 | 598.51 | ≤500 Da |
| LogP | 4.57 | 4.33 | 5.30 | 4.53 | 7.65 | 6.47 | <5 |
| Rotatable Bonds | 9 | 7 | 7 | 9 | 4 | 8 | <10 |
| HBA | 6 | 4 | 8 | 5 | 3 | 7 | ≤10 |
| HBD | 1 | 2 | 2 | 3 | 4 | 4 | ≤5 |
| Water solubility | −5.585 | −5.078 | −3.182 | −4.998 | −2.892 | −2.905 | <−10 insoluble to <0 highly soluble |
| Caco-2 permeability | 0.585 | 1.169 | 1.101 | 0.564 | 0.754 | −0.526 | >0.90 |
| IA (human) | 94.26 | 100 | 97.45 | 82.54 | 100 | 64.80 | >30 |
| Skin permeability | −2.688 | −2.802 | −2.735 | −2.752 | −2.735 | −2.735 | >−2.5 |
| P-gp substrate | Yes | Yes | No | Yes | Yes | No | No |
| P-gp I inhibitor | Yes | Yes | Yes | Yes | No | No | No |
| BBB permeability | −0.862 | −0.854 | −0.633 | −1.136 | −0.941 | −2.352 | >0.3 high to <−1 poor |
| CYP2D6 inhibitor | No | No | No | No | No | No | No |
| hERG I inhibitor | No | No | No | No | Yes | No | No |
| Total clearance | 0.544 | 0.152 | −0.023 | −0.124 | 0.813 | −0.171 | <0.3 low to >0.7 high |
| Renal OCT2 substrate | No | No | No | No | Yes | No | No |
| Compound Name | IUPAC Name | Molecular Structure |
|---|---|---|
| Natural Compound Hits | ||
| Hit1 | N-(4-(furan-2-yl)butan-2-yl)-2-((3-(4-methoxyphenyl)-4-oxo-4H-chromen-7-yl)oxy)acetamide |  |
| Hit2 | (S)-4-(8-methoxy-11b-methyl-1,3-dioxo-5,6-dihydro-1H-imidazo[1 ‘,5′:1,2]pyrido[3,4-b]indol-2(3H,11H,11bH)-yl)-N-pentylbenzamide |  |
| Synthetic Compound Hits | ||
| Hit1 | 4-((4-(4-methoxyphenyl)phthalazin-1-yl)amino)-N-(thiazol-2-yl)benzenesulfonamide |  |
| Hit2 | (E)-N-(1-(5-(2,5-dichlorophenyl)furan-2-yl)-3-((3-hydroxypropyl)amino)-3-oxoprop-1-en-2-yl)-4-methoxybenzamide |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parate, S.; Kumar, V.; Danishuddin; Hong, J.C.; Lee, K.W. Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 5311. https://doi.org/10.3390/ijms22105311
Parate S, Kumar V, Danishuddin, Hong JC, Lee KW. Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics. International Journal of Molecular Sciences. 2021; 22(10):5311. https://doi.org/10.3390/ijms22105311
Chicago/Turabian StyleParate, Shraddha, Vikas Kumar, Danishuddin, Jong Chan Hong, and Keun Woo Lee. 2021. "Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics" International Journal of Molecular Sciences 22, no. 10: 5311. https://doi.org/10.3390/ijms22105311
APA StyleParate, S., Kumar, V., Danishuddin, Hong, J. C., & Lee, K. W. (2021). Computational Investigation Identified Potential Chemical Scaffolds for Heparanase as Anticancer Therapeutics. International Journal of Molecular Sciences, 22(10), 5311. https://doi.org/10.3390/ijms22105311