
Hydrogen Bonds: Raman Spectroscopic Study

Abstract
:1. Introduction
2. Definition, Brief History, Main Stages of Development
3. General Description
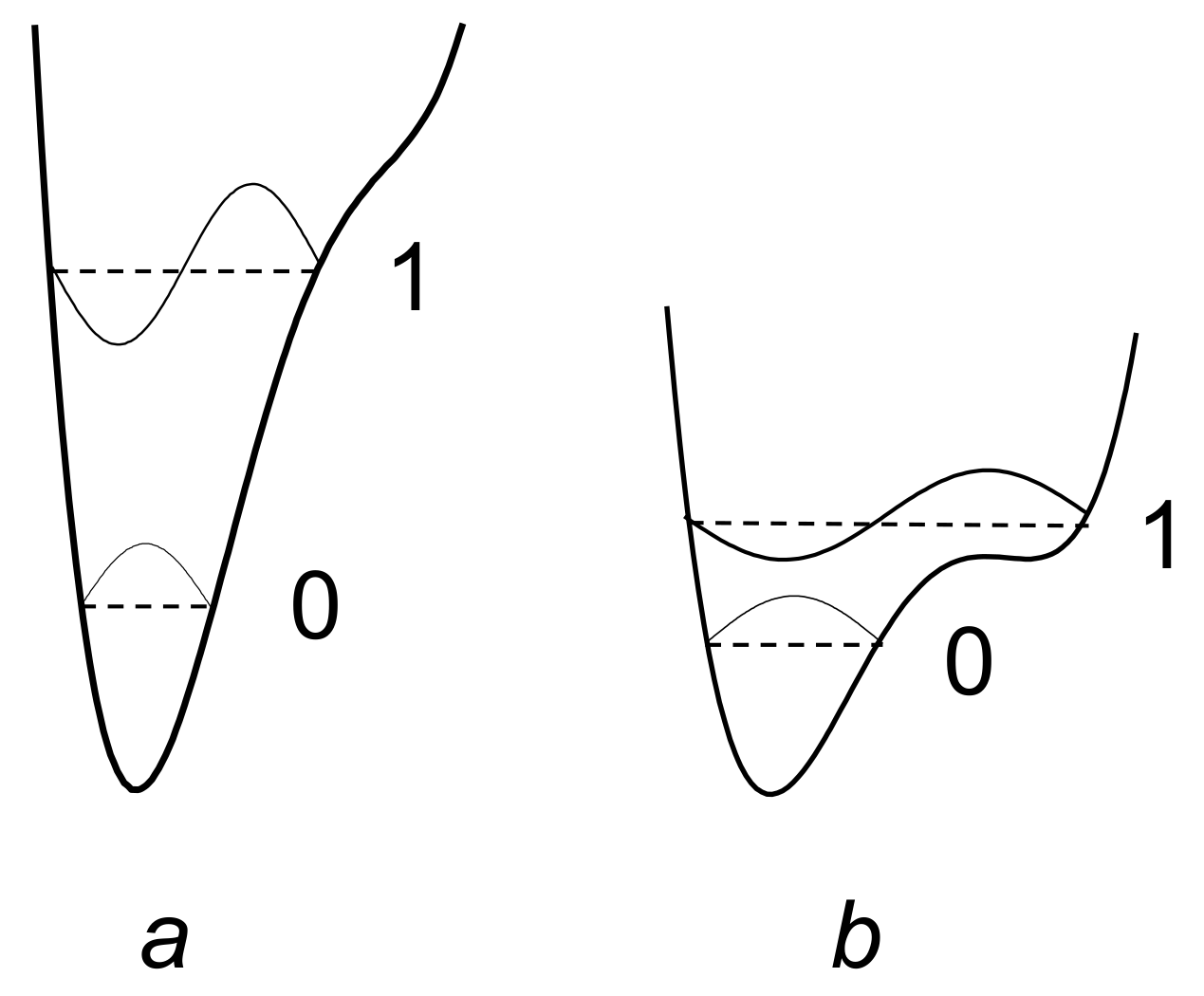
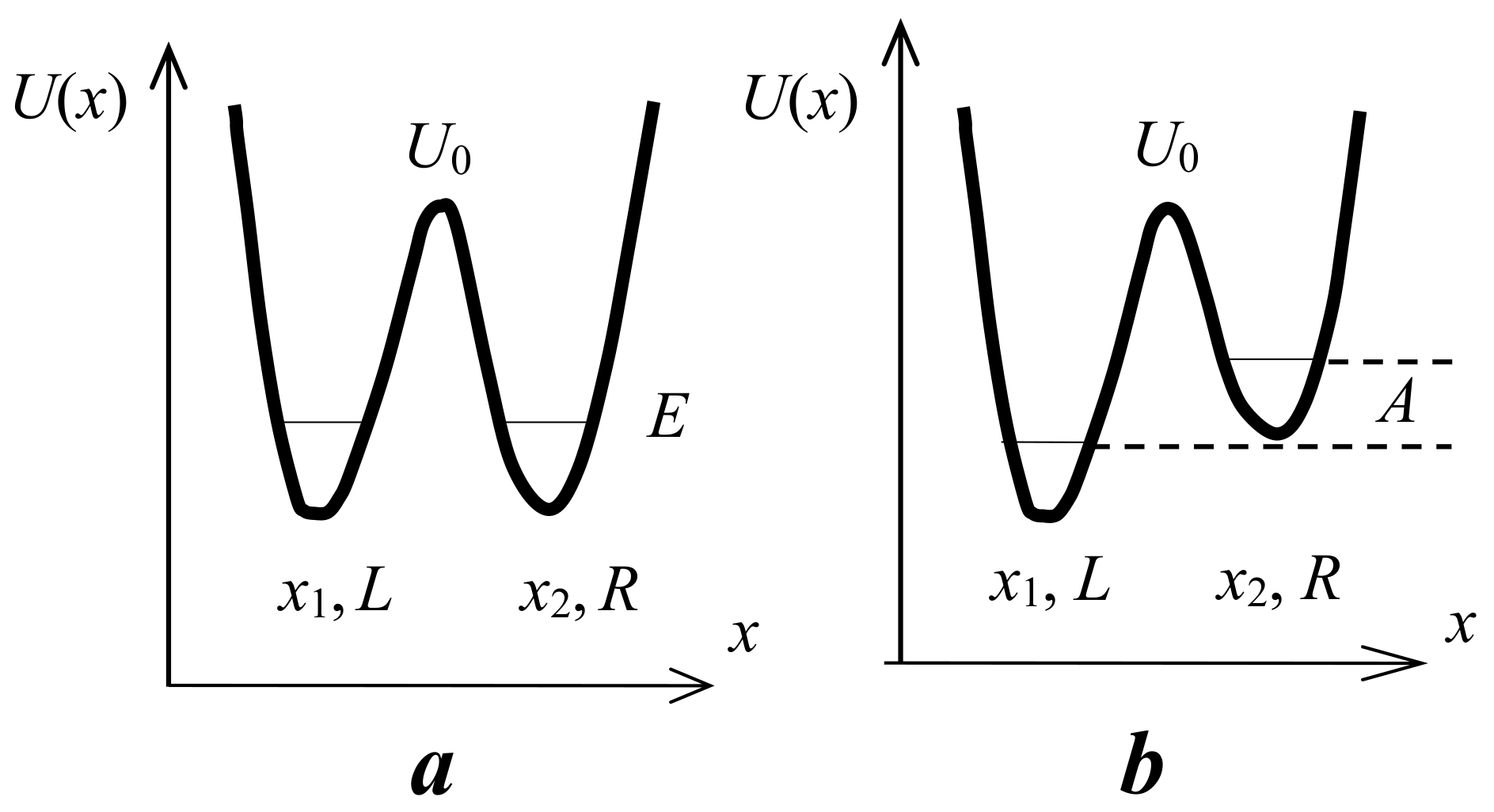
3.1. Potential Energy of a Proton on a Hydrogen Bond
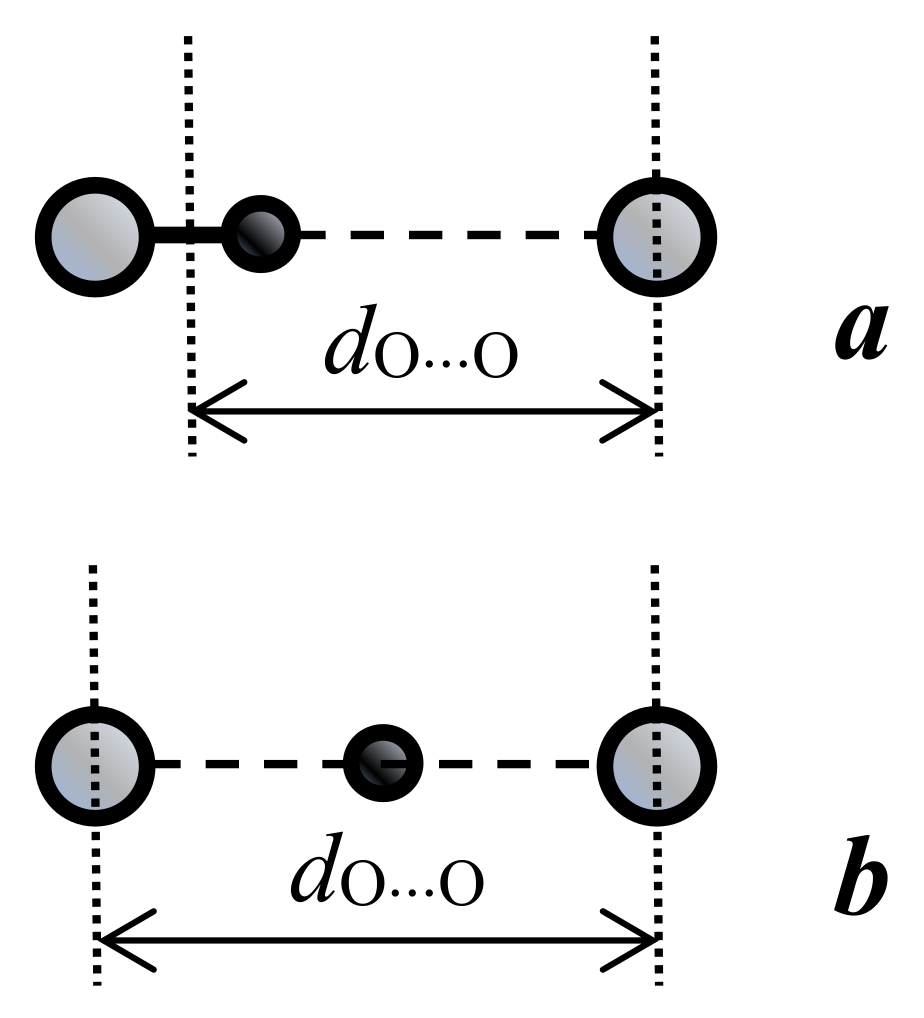
3.2. The Bond Energy as Function of its Length. Uncertainty of the Proton Coordinates, Uncertainty of the O···O Distance
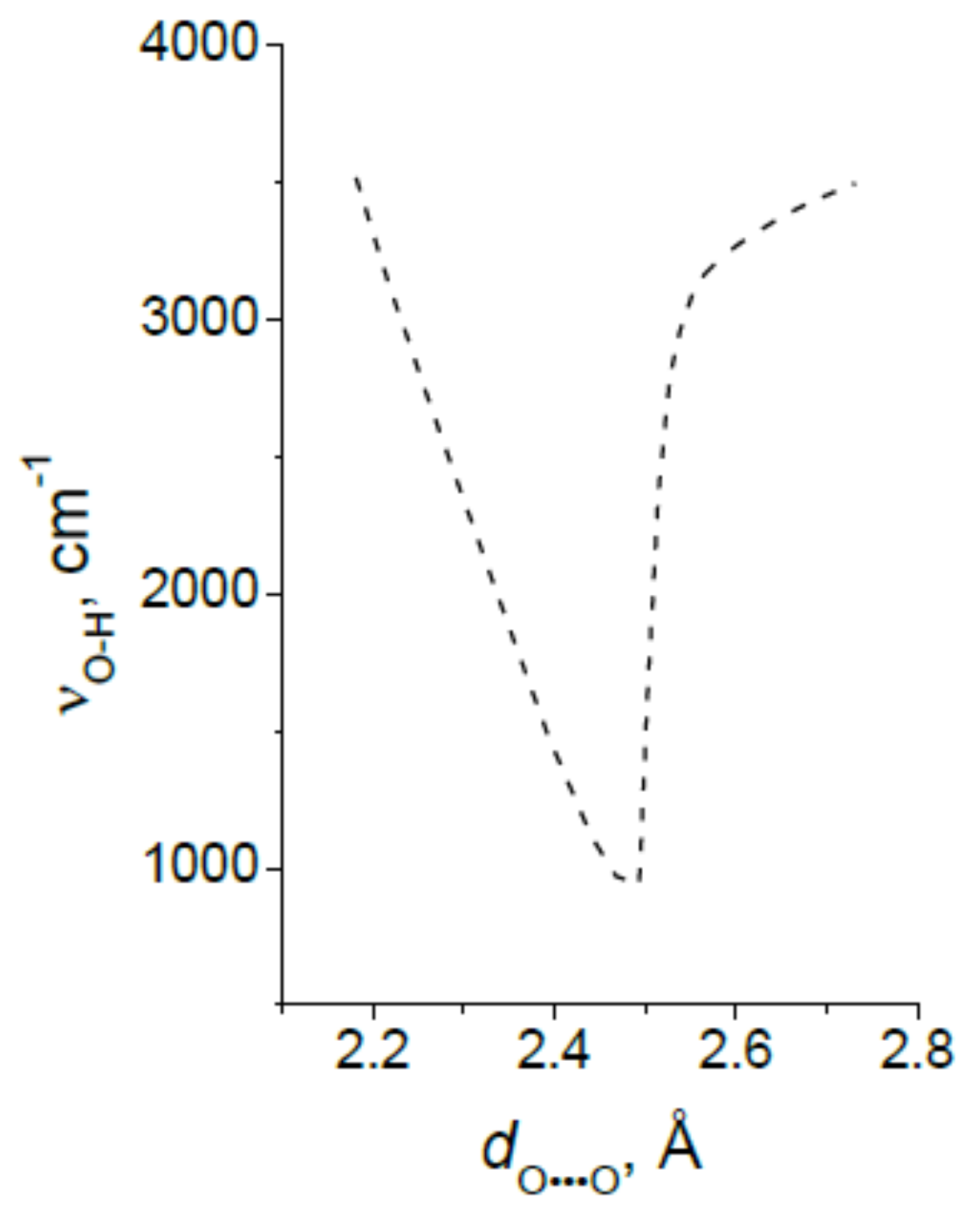
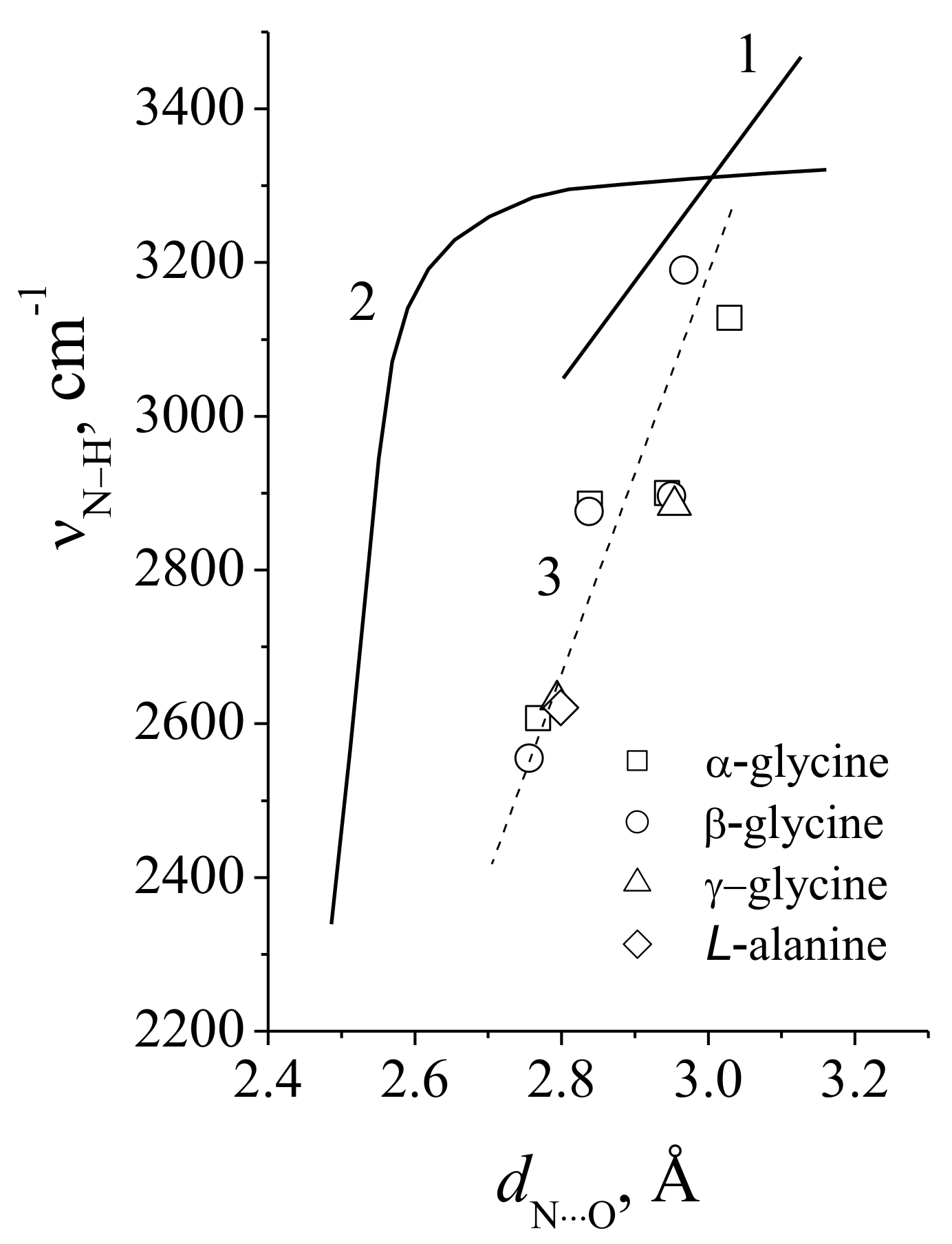
3.3. The Proton Vibrational Frequency as Function of a Length of the Hydrogen Bond
3.4. Half-Width of the O–H···O Vibrational Bands
3.5. Intensity of O–H···O Vibrational Bands
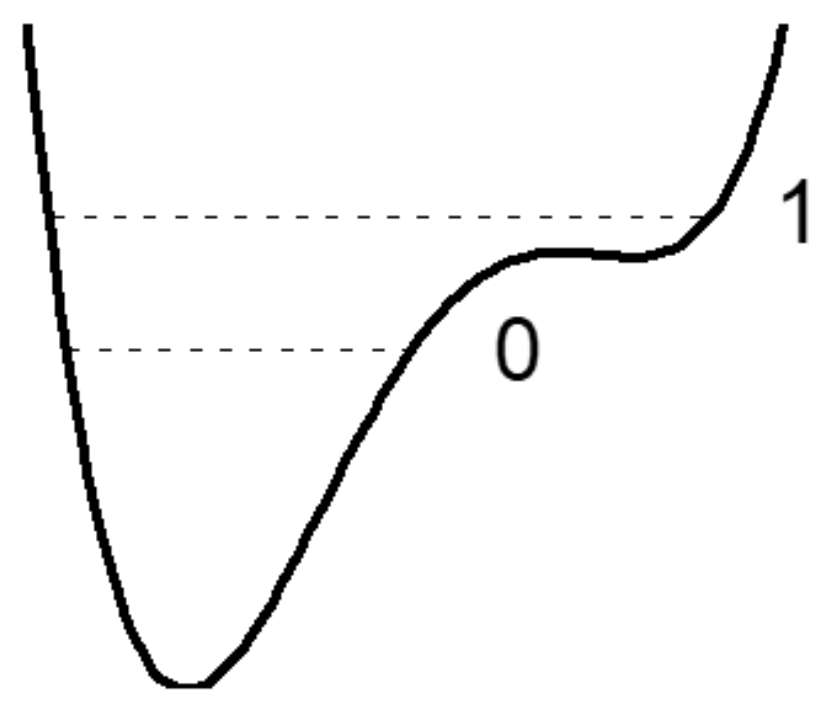
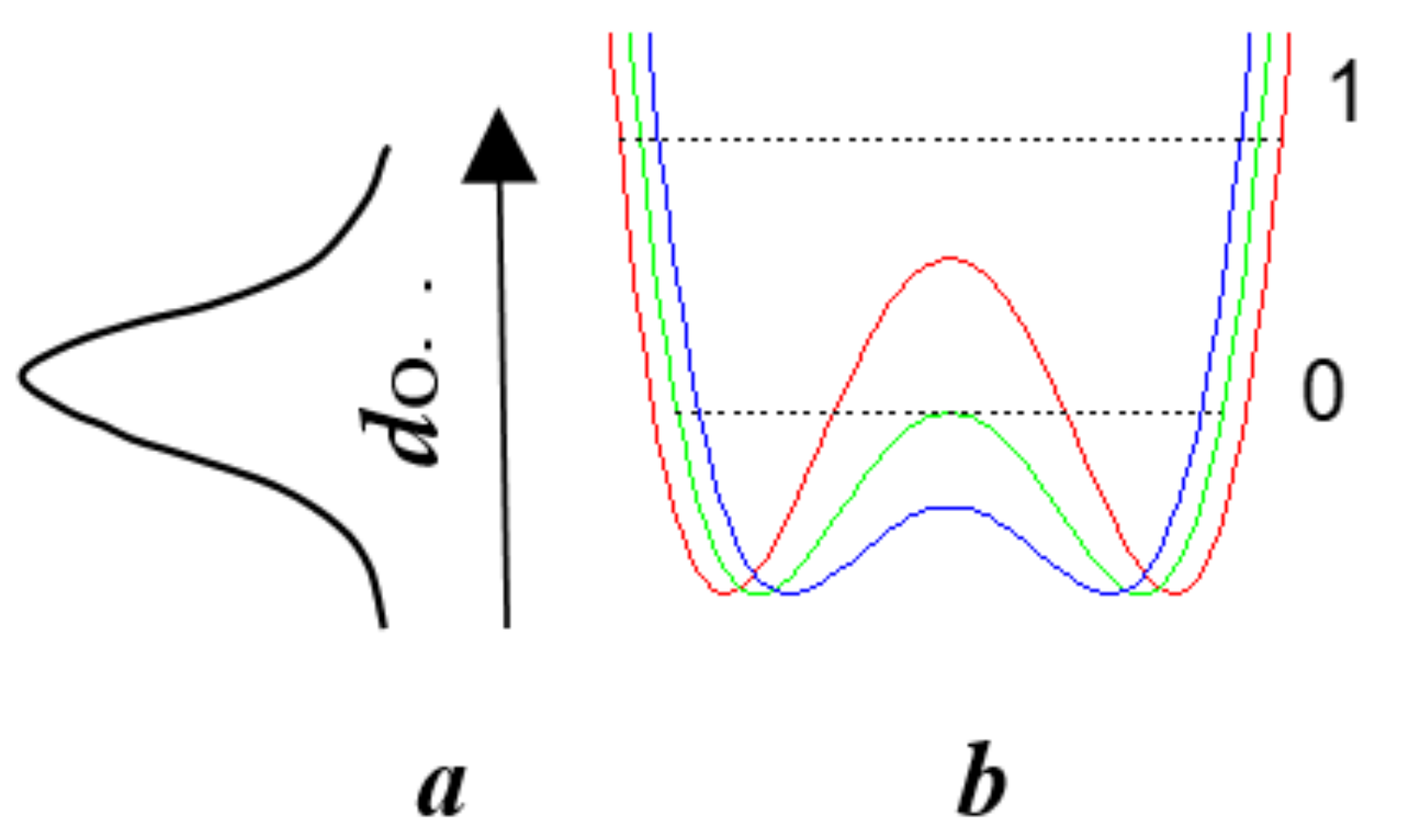
3.6. How Does Single-Well Potential Occur?
- Identity of a donor and an acceptor, which in practice simply means the formation of a hydrogen bond by two identical molecules. If the molecules are different, then the interaction of the proton with the donor will always be stronger than with the acceptor, and the O–H length is shorter than H···O. It prevents the formation of the minimum dO···O and strong hydrogen bond. When the molecules are completely identical, the concepts “donor” and “acceptor” lose their meaning.
- Electronegativity of oxygen-donor and oxygen-acceptor. It was already mentioned above that electronegativity determines how strongly an oxygen atom interacts with a proton, i.e., how close a proton can come to an oxygen atom, both a donor and an acceptor.
- Thermal vibrations. Thermal vibrations increase the distance between atoms due to anharmonic processes. In a molecular crystal, the vibrational spectrum starts from 20–40 cm−1. Consequently, the excitation of equilibrium thermal vibrations starts from 40–60 K. In other words, the minimum possible hydrogen bond length can be established only at T ≤ 40–60 K.
4. Experimental Study of Strong Hydrogen Bonds
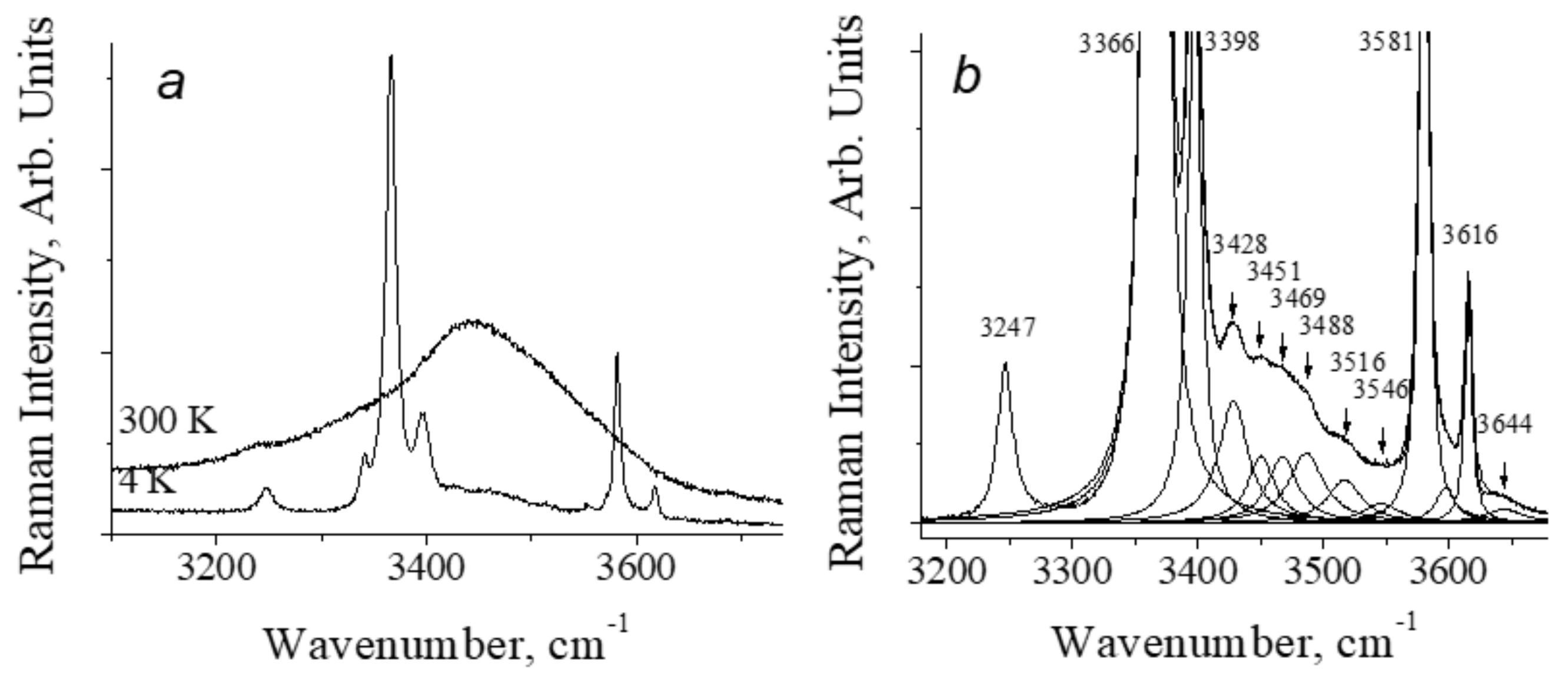
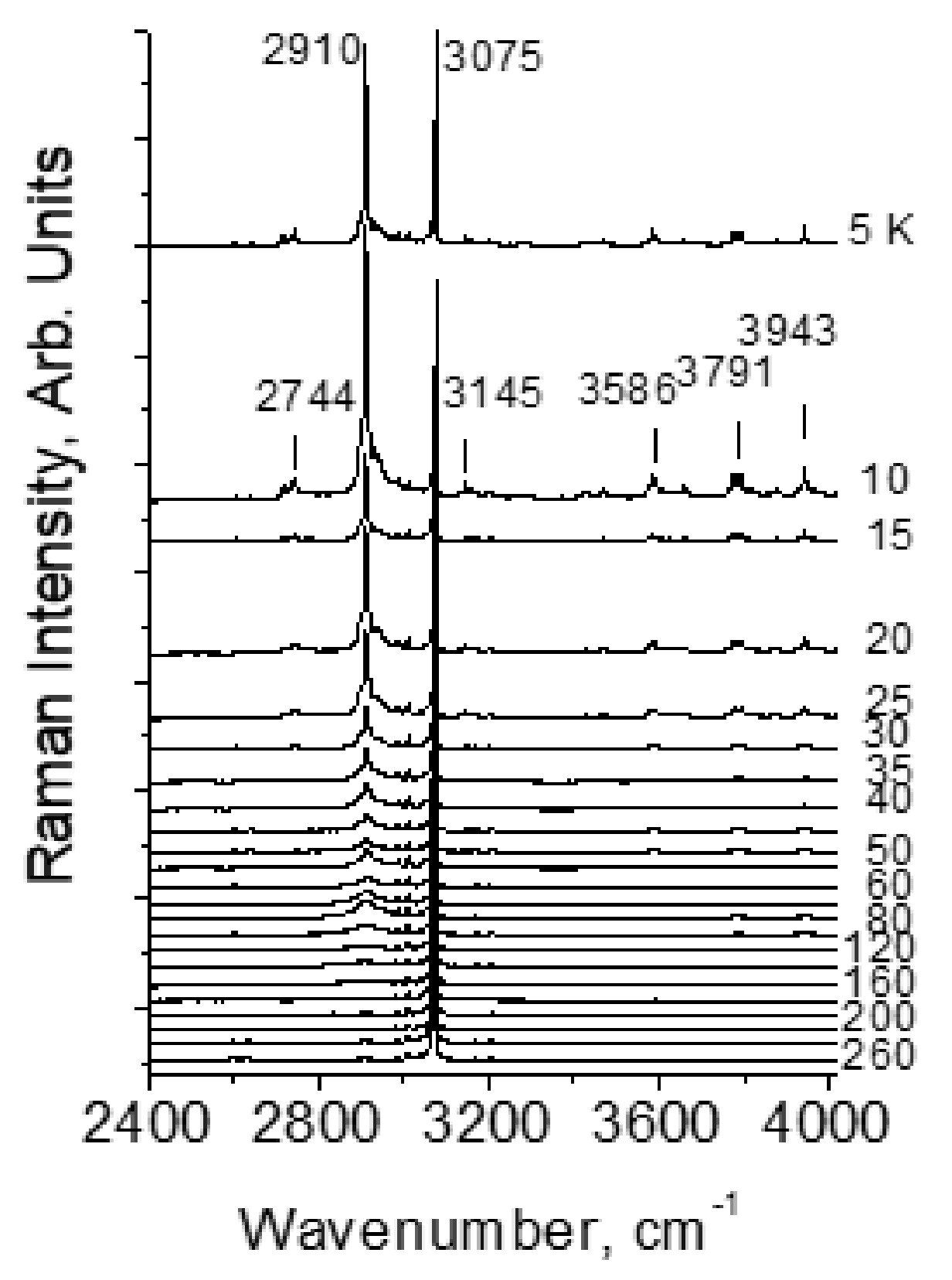
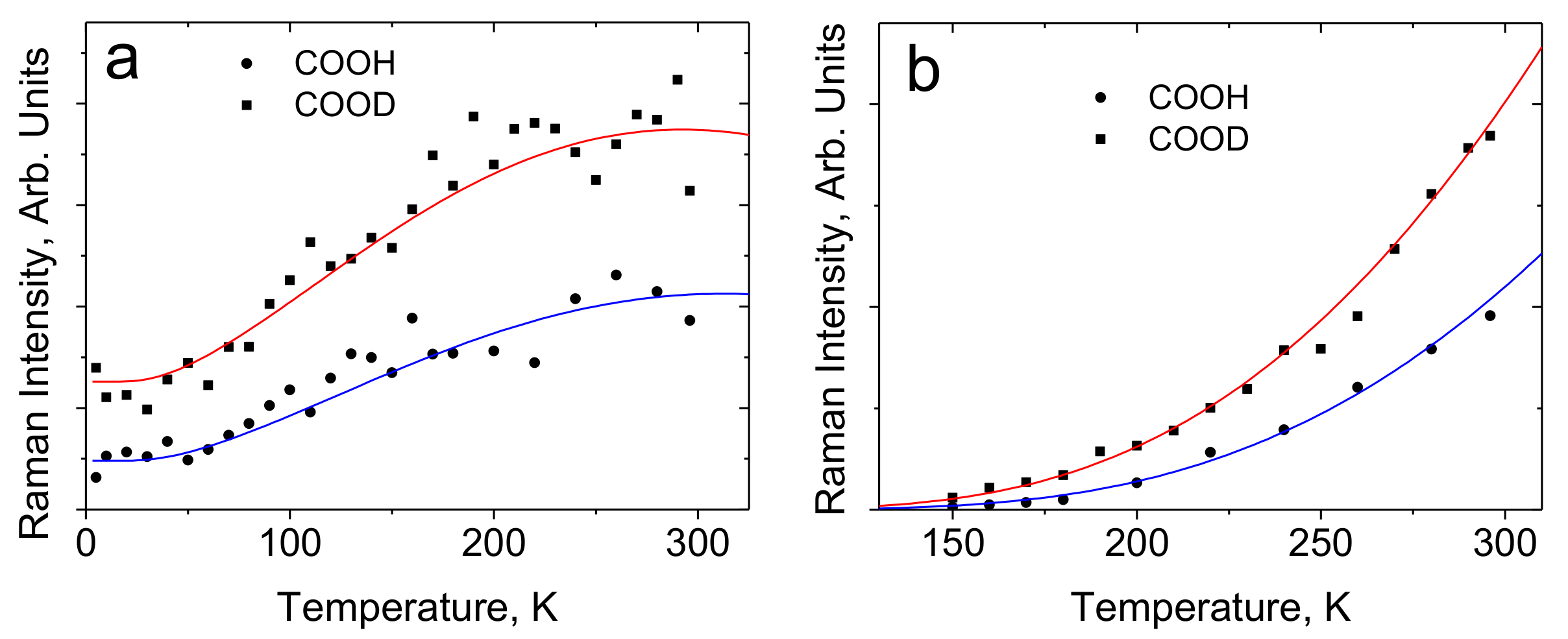
4.1. The Features of the Vibrational Spectrum of Benzoic Acid
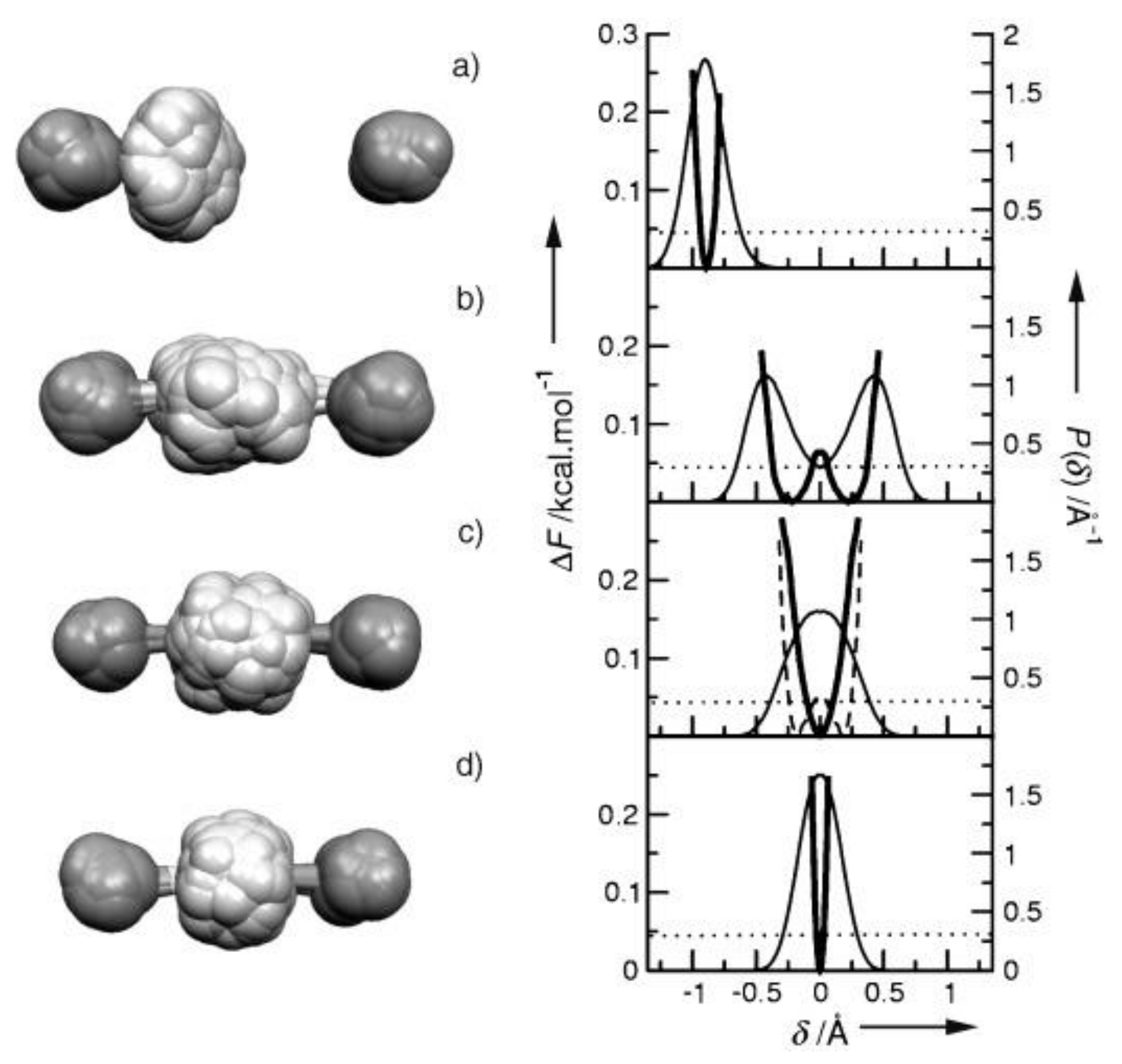
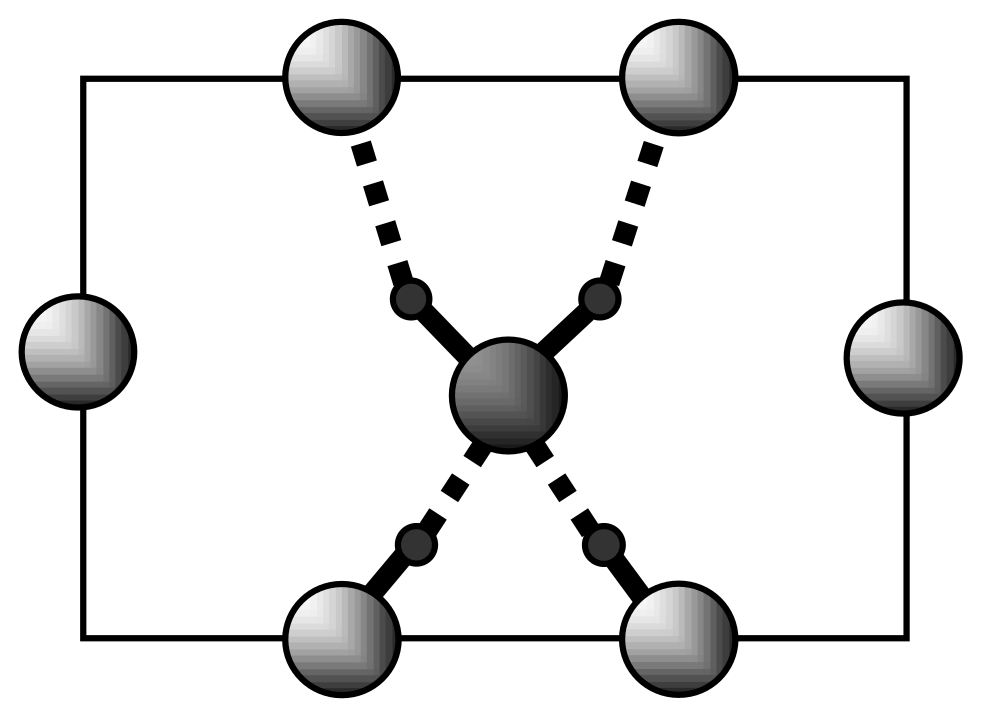
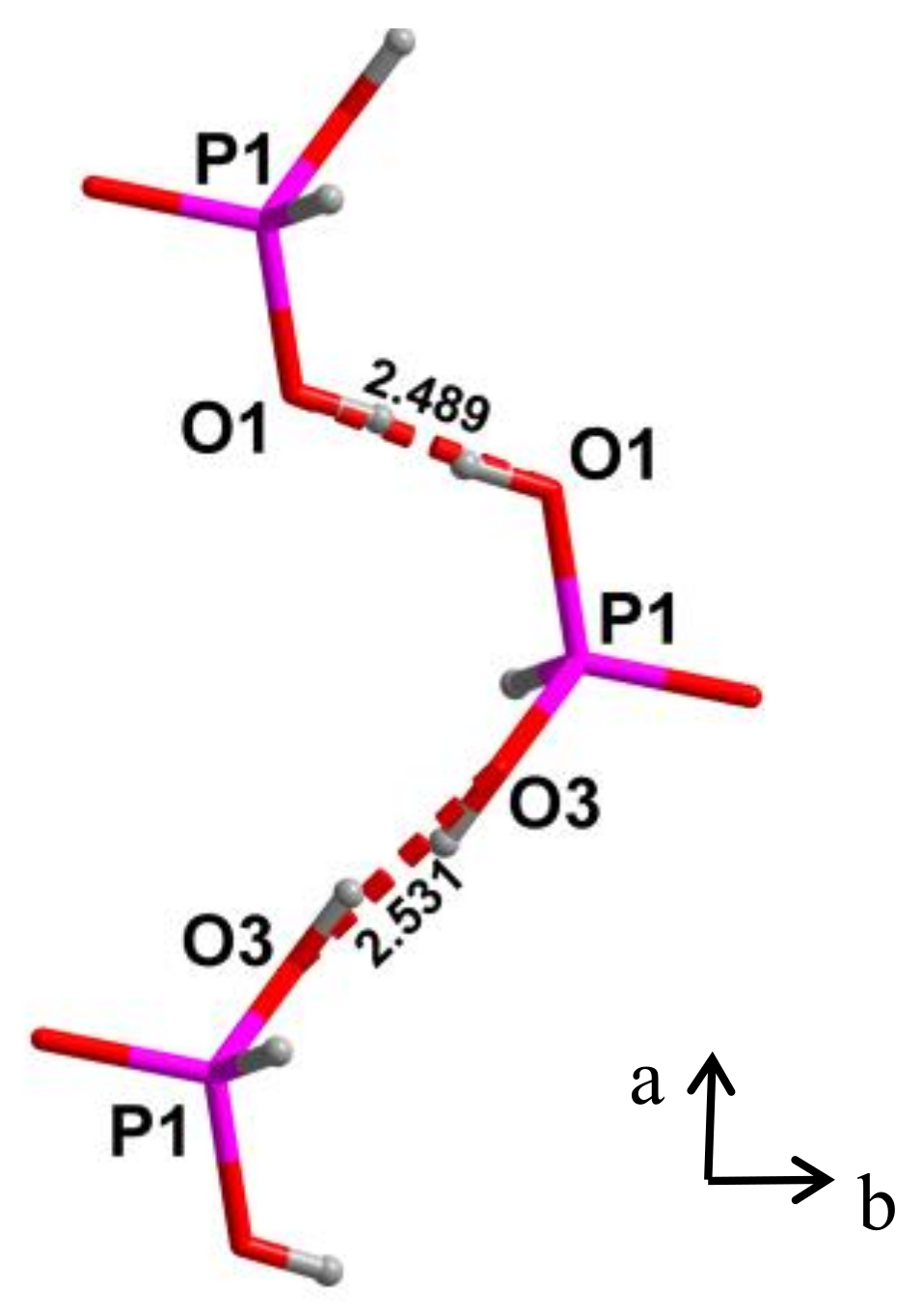
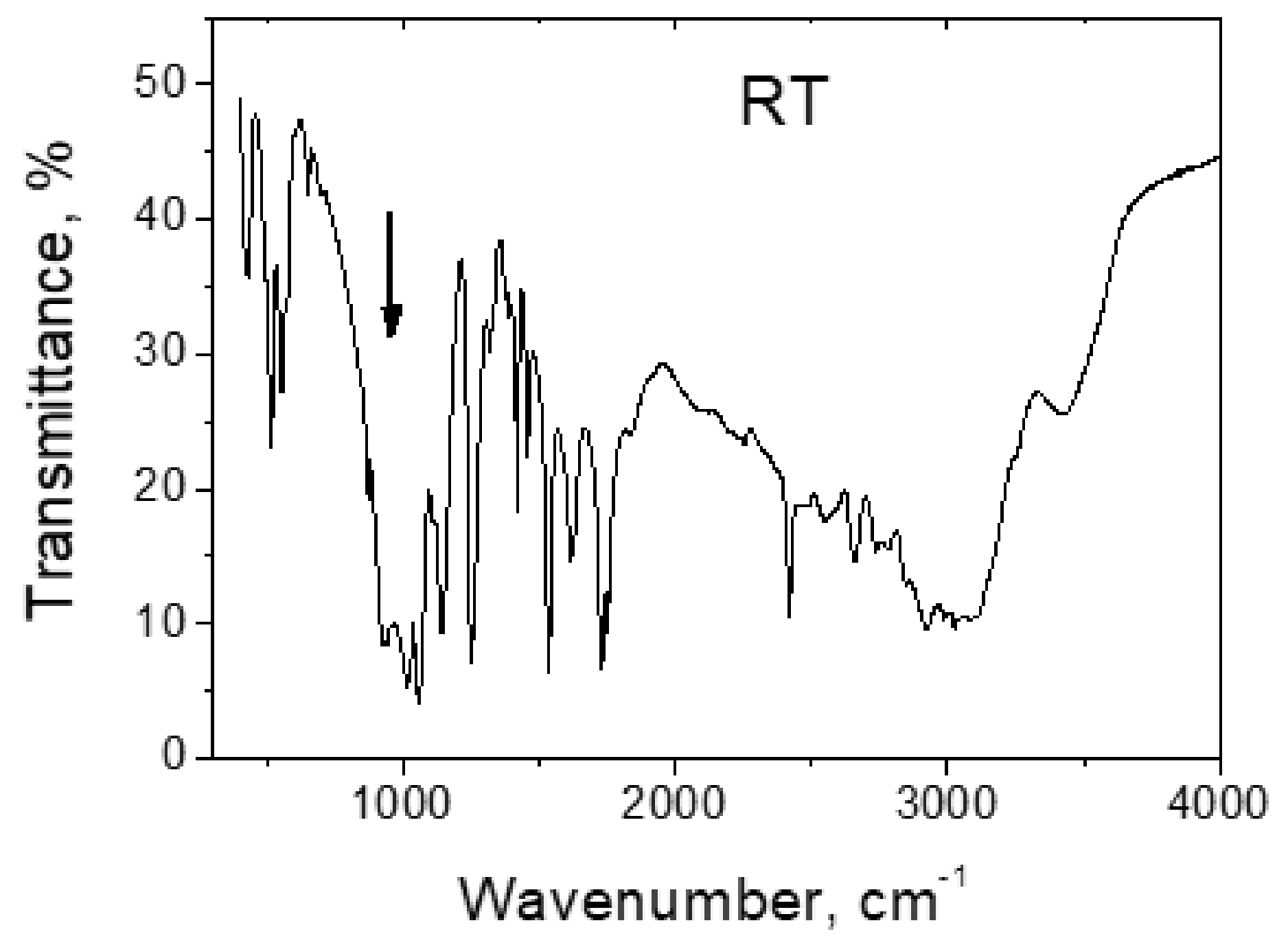
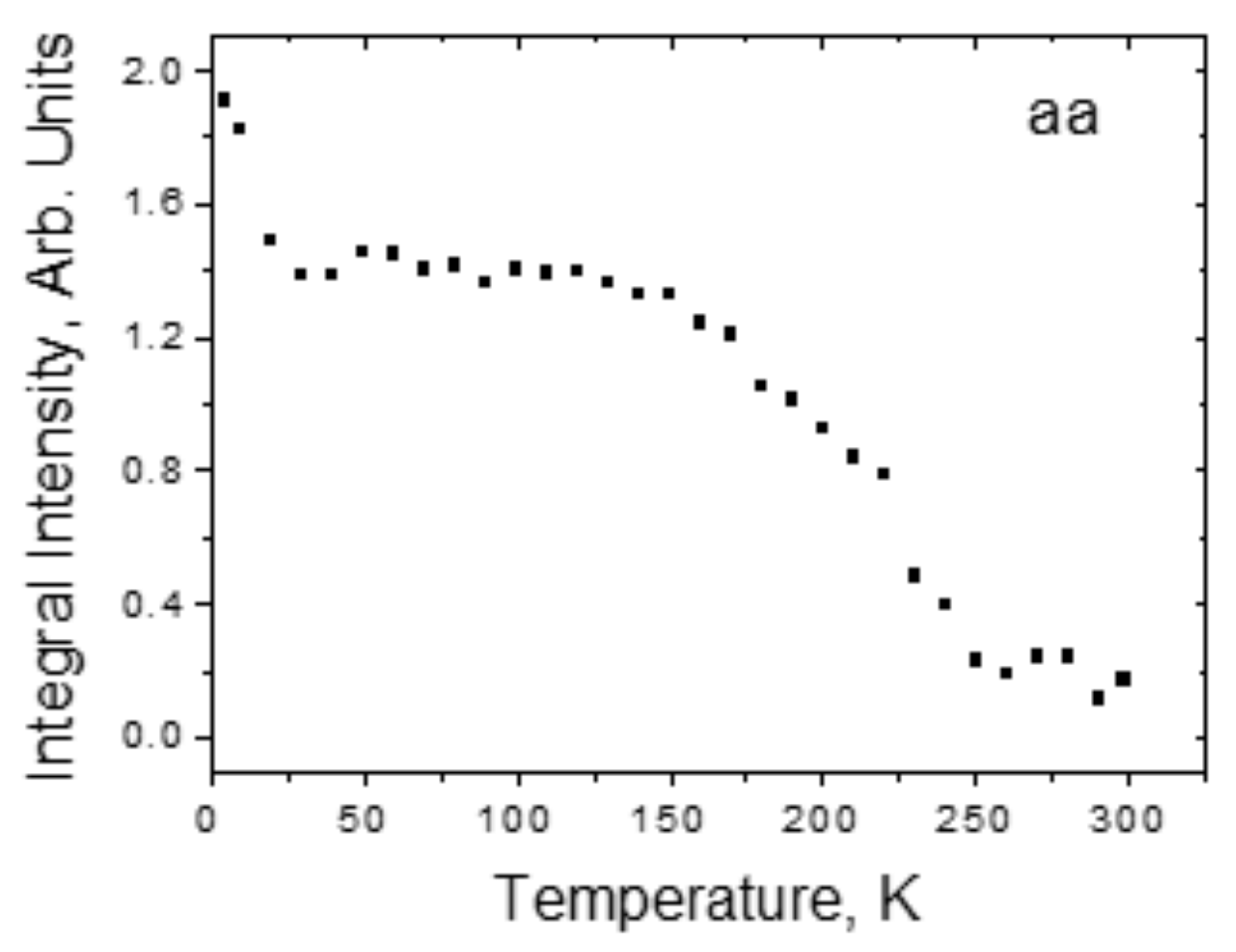
4.2. Strong Hydrogen Bonds. Glycine Phosphate
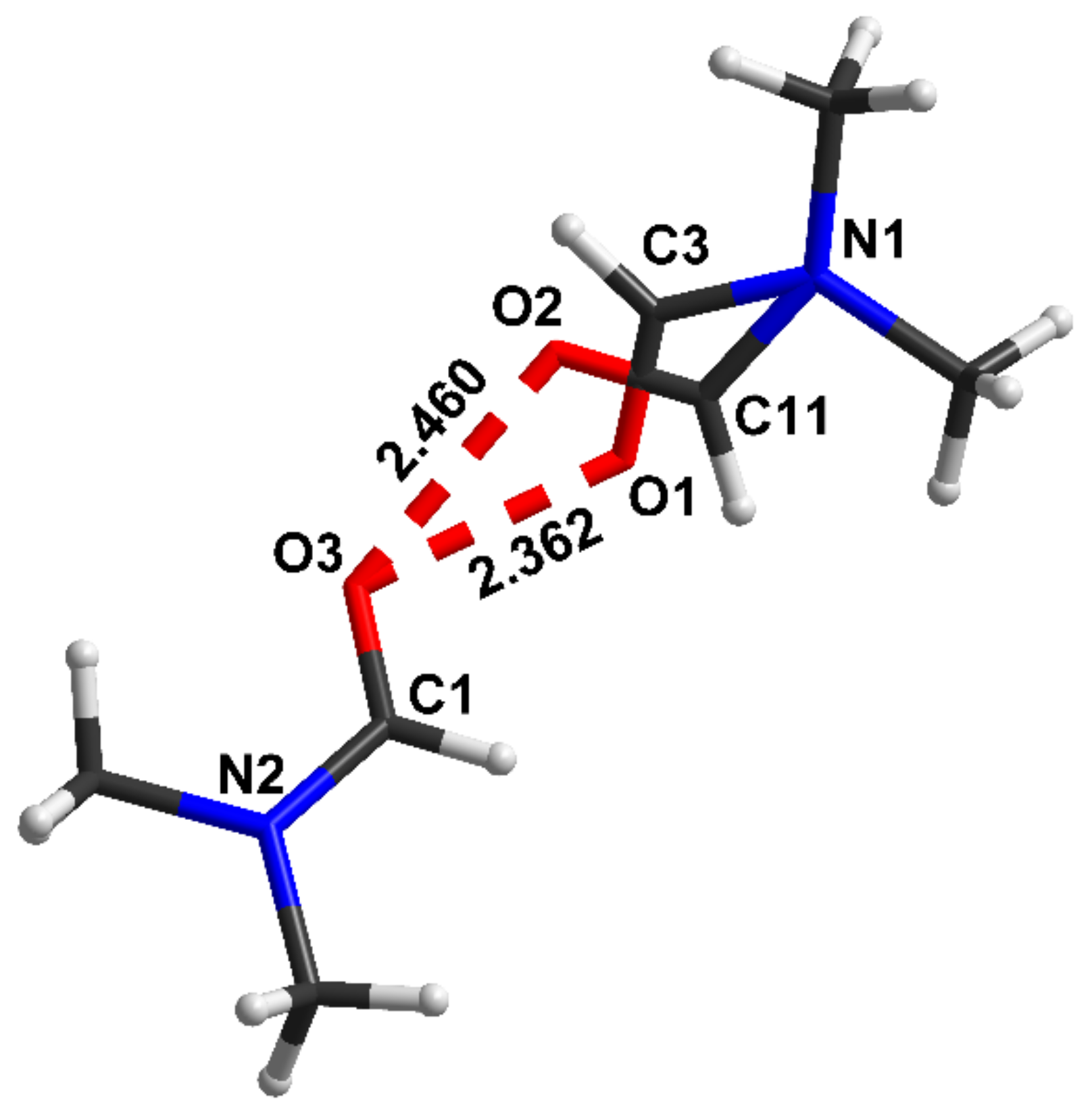
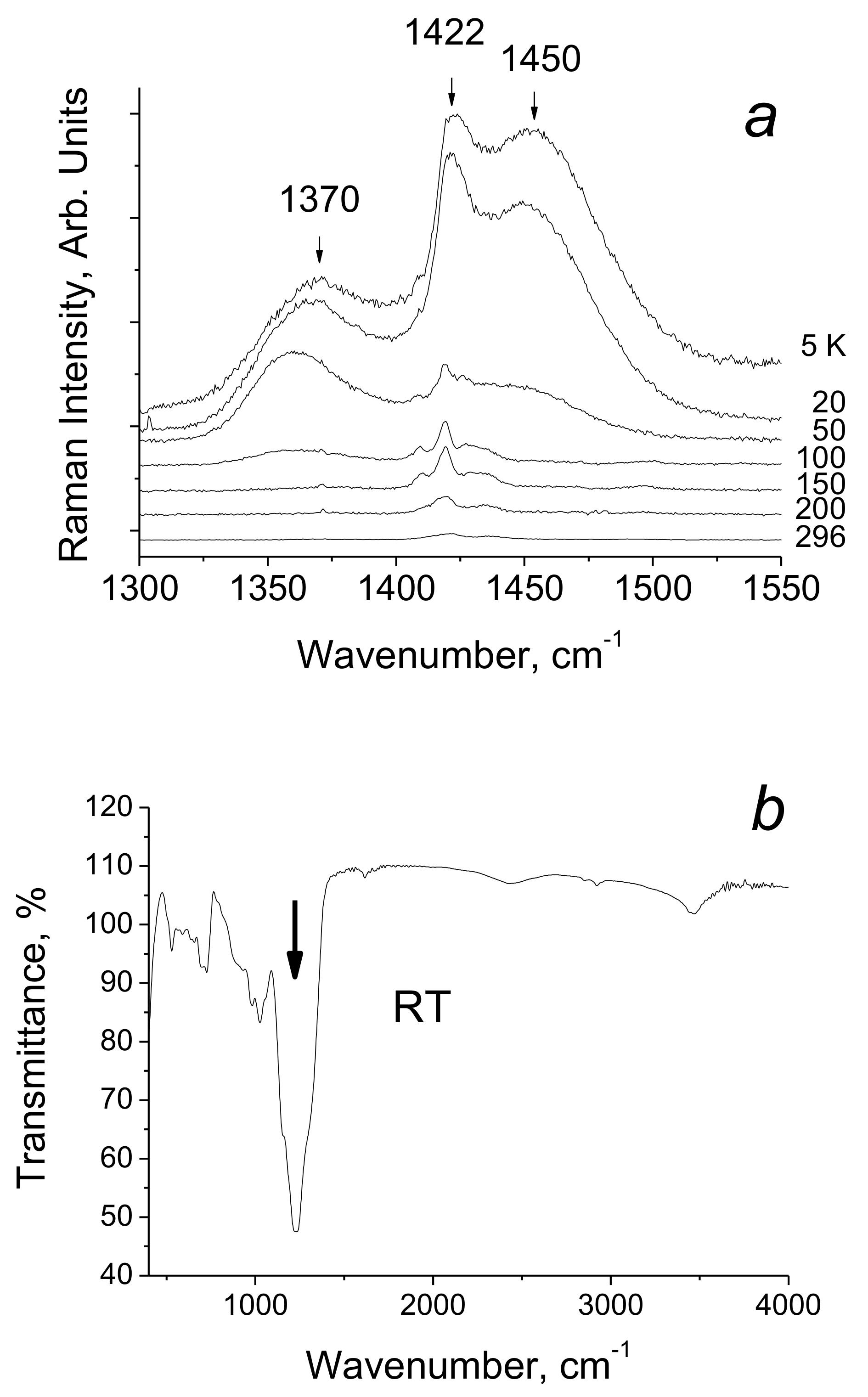
4.3. Extremely Strong Hydrogen Bond in [(DMF)2H]2
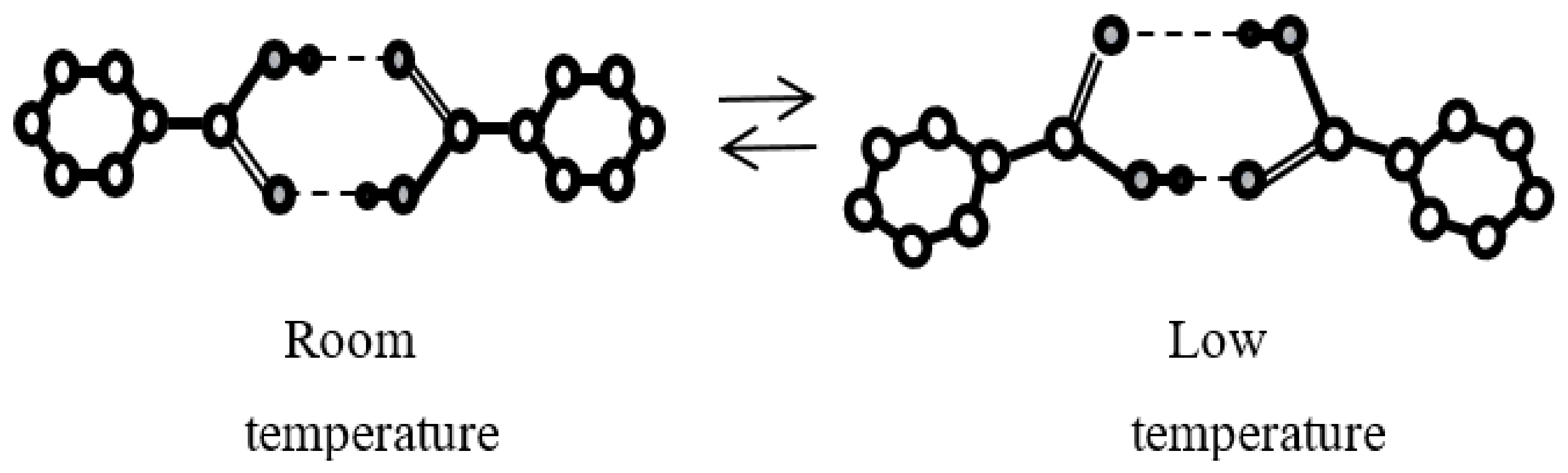
5. Tautomeric Hydrogen Bonds
5.1. What Is the Proton Tautomerism?
- Coordinated proton tunneling on the τ-bond occurs mainly at low temperatures, and proton hopping occurs mainly at high temperatures;
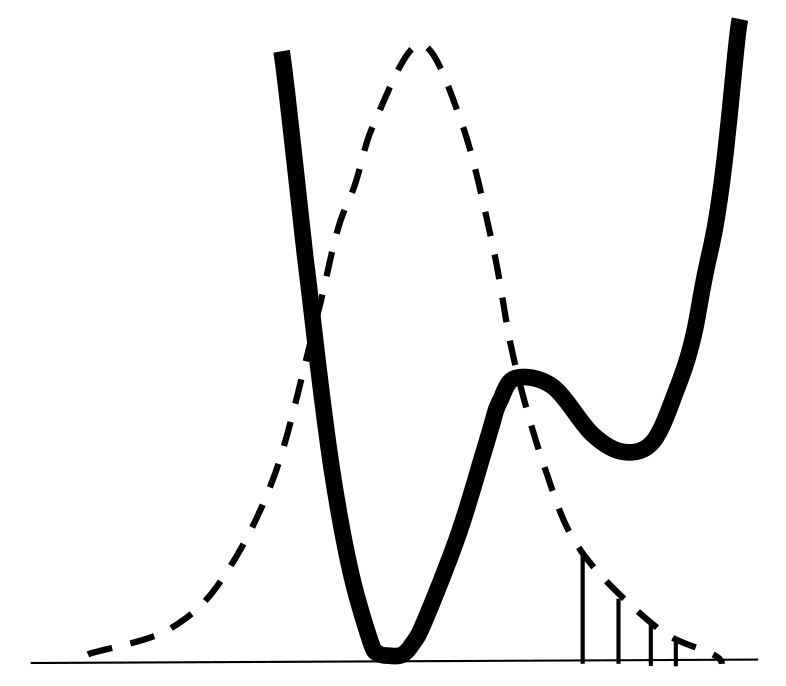
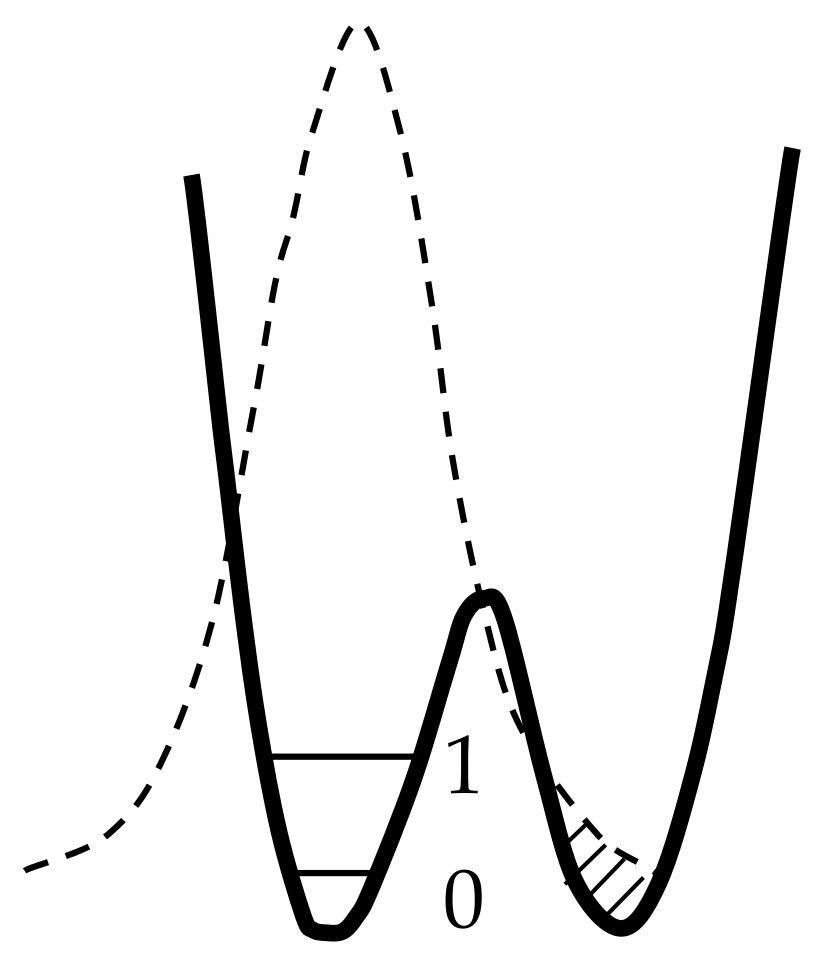
- Tunneling changes the energy of their interaction with the environment and requires the participation of phonons ωA (Figure 24b);
- Spread of proton (deuteron) distribution function to the neighboring well increases the hydrogen τ-bond (Figure 24a);
- Proton tunneling and proton hopping do not change the force constant of the τ-bond but modulate the length of C–O and C = O bonds of the τ-ring;
- Deuteration of the τ-bond virtually does not affect the degree of proton sharing and significantly slows down the tunneling.
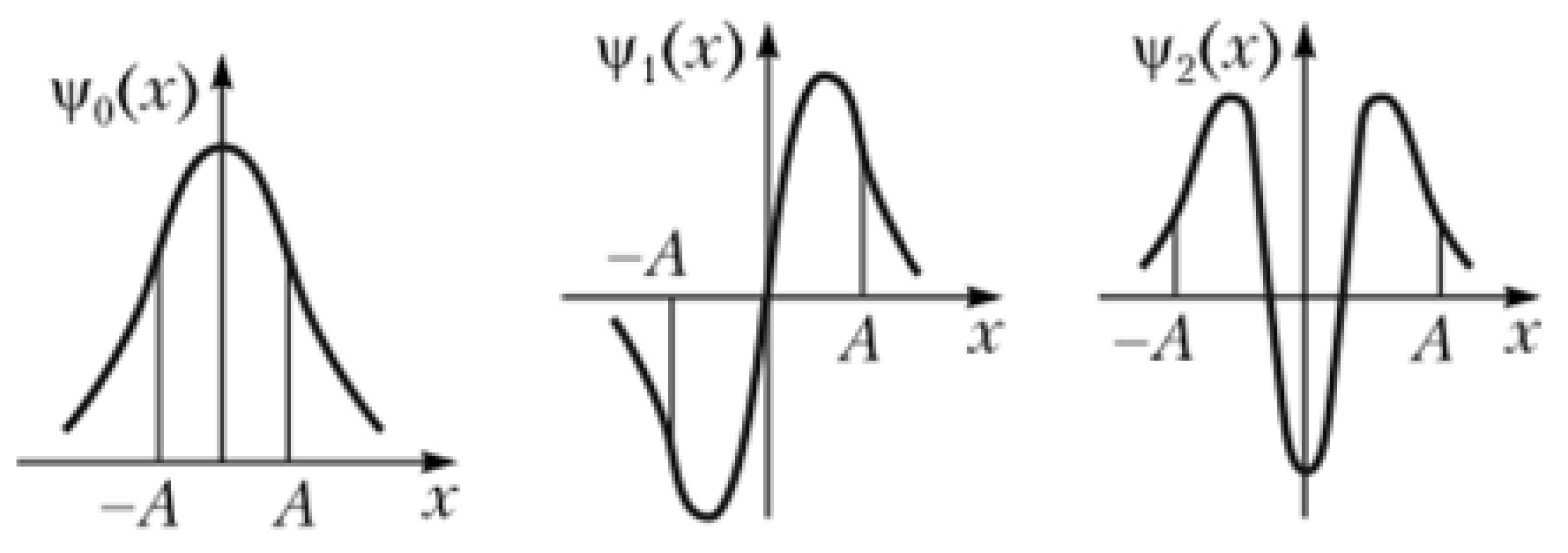
5.2. Quantum Delocalization of Protons
5.3. Proton Hopping in Ibuprofen
5.4. Proton Tunneling
6. Brief Characteristic of N–H···O and C–H···Y Hydrogen Bonds
6.1. N–H···O Hydrogen Bond
6.2. Weak C–H···Y Bonds. “Blue” Shift
7. Conclusions
8. Experimental
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hadzi, D. (Ed.) The Hydrogen Bond; Pergamon Press: New York, NY, USA; London, UK, 1957. [Google Scholar]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Hamilton, W.C.; Ibers, J.A. Hydrogen Bonding in Solids; Benjann: New York, NY, USA, 1968. [Google Scholar]
- Vinogradov, S.N.; Linnell, R.H. Hydrogen Bonding; Van Nostrand Reinhold: New York, NY, USA, 1971. [Google Scholar]
- Joesten, M.D.; Schaad, L.J. Hydrogen Bonding; Dekker: New York, NY, USA, 1974. [Google Scholar]
- Kollman, P.A.; Allen, L.C. The theory of the hydrogen bond. Chem. Rev. 1972, 72, 283–303. [Google Scholar] [CrossRef]
- Kollman, P.A. A general analysis of noncovalent intermolecular interaction. J. Am. Chem. Soc. 1977, 99, 4875–4893. [Google Scholar] [CrossRef]
- Morokuma, K. Why do molecules interact? The origin of electron donor acceptor complexes, hydrogen bonding and proton affinity. Acc. Chem. Res. 1977, 10, 294–300. [Google Scholar] [CrossRef]
- Schuster, P.; Zundel, G.; Sandorfy, C. The Hydrogen Bond. Recent Developments in Theory and Experiment; North Holland: Amsterdam, The Netherlands, 1976; Volume 3. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA; Oxford, UK, 1997. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Huggins, M.L. The Role of Hydrogen Bonds in Conduction. Diploma Thesis, University of California, Berkeley, CA, USA, 1919. [Google Scholar]
- Latimer, W.M.; Rodebush, W.H. Polarity and ionization from the standpoint of the Lewis theory of valence. J. Am. Chem. Soc. 1920, 42, 1419. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Bernal, J.D.; Fowler, R.H. Theory of water and ionic solution with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1933, 1, 515. [Google Scholar] [CrossRef]
- Morokuma, K. Molecular orbital studies of hydrogen bonds. J. Chem. Phys. 1971, 55, 1236–1244. [Google Scholar] [CrossRef]
- Benoit, M.; Marx, D. The Shapes of Protons in Hydrogen Bonds Depend on the Bond Length. Chem. Phys. Chem. 2005, 6, 1738–1741. [Google Scholar] [CrossRef]
- Wang, L.; Fried, S.D.; Boxer, S.G.; Markland, T.E. Quantum delocalization of protons in the hydrogen-bond network of an enzyme active site. Proc. Natl. Acad. Sci. USA 2014, 111, 18454. [Google Scholar] [CrossRef] [Green Version]
- Goncharov, A.F.; Struzhkin, V.V.; Somayazulu, M.S.; Hemley, R.J.; Mao, H.K. Compression of Ice to 210 Gigapascals: Infrared Evidence for a Symmetric Hydrogen-Bonded Phase. Science 1996, 273, 218–220. [Google Scholar] [CrossRef]
- Aoki, K.; Yamawaki, H.; Sakashita, M.; Fujihisa, H. Infrared absorption study of the hydrogen-bond symmetrization in ice to 110 GPa. Phys. Rev. B 1996, 54, 15673–15677. [Google Scholar] [CrossRef]
- Novak, A. Hydrogen bonding in solids. Correlation of spectroscopic and crystallographic data. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1974; Volume 18, pp. 177–216. [Google Scholar]
- Kolesov, B.A. Raman investigation of H2O molecule and hydroxyl groups in the channels of hemimorphite. Am. Miner. 2006, 91, 1355–1362. [Google Scholar] [CrossRef]
- Galkina, Y.A.; Kryuchkova, N.A.; Vershinin, M.A.; Kolesov, B.A. Features of strong O–H⋯ O and N–H⋯ O hydrogen bond manifestation in vibrational spectra. J. Struct. Chem. 2017, 58, 911–918. [Google Scholar] [CrossRef]
- Kolesov, B.A. Unusual behavior of benzoic acid at low temperature: Ramanspectroscopic study. Spectrochim. Acta Part A 2015, 142, 320–323. [Google Scholar] [CrossRef]
- Averbuch-Pouchot, M.-T. Structures of Glycinium Phosphite and Glycylglycinium Phosphite. Acta Cryst. 1993, C49, 815–818. [Google Scholar] [CrossRef]
- Shikanai, F.; Komukae, M.; Czapla, Z.; Osaka, T. Crystal Structure of NH3CH2COOHH2PO3 in the Ferroelectric Phase. J. Phys. Soc. Jpn. 2002, 71, 498–503. [Google Scholar] [CrossRef]
- Shikanai, F.; Yamasaki, M.; Komukae, M.; Osaka, T. Structural Study of Partially Deuterated Glycinium Phosphite in the Paraelectric Phase. J. Phys. Soc. Jpn. 2003, 72, 325–329. [Google Scholar] [CrossRef]
- Machida, M.; Uchida, H.; Ishibashi, T.; Taniguch, H.; Komukae, M.; Osaka, T.; Koyano, N. Neutron Diffraction Study of Crystal Structures of Deuterated Glycinium Phosphite in Paraelectric and Ferroelectric Phases. J. Phys. Soc. Jpn. 2004, 73, 107–115. [Google Scholar] [CrossRef]
- Perumal, R.; Senthil Kumar, K.; Moorthy Babu, S.; Bhagavannarayana, G. Optical characterization of ferroelectric glycinium phosphite single crystals. J. Alloy. Compd. 2010, 490, 342–349. [Google Scholar] [CrossRef]
- Kolesov, B.A.; Chupina, A.V.; Berezin, A.S.; Kompankov, N.B.; Abramov, P.A.; Sokolov, M.N. Proton motion inside [(DMF)2H]2[W6Cl14]: Structural, Raman and luminescence studies. Phys. Chem. Chem. Phys. 2020, 22, 25344–25352. [Google Scholar] [CrossRef]
- Foces-Foces, C.; Echevarría, A.; Jagerovic, N.; Alkorta, I.; Elguero, J.; Langer, U.; Klein, O.; Minguet-Bonveh, M.; Limbach, H.-H. A Solid-State NMR, X-ray Diffraction, and ab Initio Computational Study of Hydrogen-Bond Structure and Dynamics of Pyrazole-4-Carboxylic Acid Chains. J. Am. Chem. Soc. 2001, 123, 7898–7906. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.; Terao, T.; Imashiro, F.; Saika, A.; Hirota, N.; Hayashi, S. A study on the proton transfer in the benzoic acid dimer by 13C high-resolution solid-state NMR and proton T1 measurements. Chem. Phys. Lett. 1981, 80, 580–584. [Google Scholar] [CrossRef]
- Meier, B.H.; Graf, F.; Ernst, R.R. Structure and dynamics of intramolecular hydrogen bonds in carboxylic acid dimers: A solid state NMR study. J. Chem. Phys. 1982, 76, 767–774. [Google Scholar] [CrossRef]
- Meier, B.H.; Meyer, R.; Ernst, R.R.; Zolliker, P.; Furrer, A.; Hälg, W. Neutron scattering study of dynamically disordered hydrogen bonds: Terephthalic acid. Chem. Phys. Lett. 1983, 103, 169–174. [Google Scholar] [CrossRef]
- Nagaoka, S.; Terao, T.; Imashiro, F.; Saika, A.; Hirota, N.; Hayashi, S. An NMR relaxation study on the proton transfer in the hydrogen bonded carboxylic acid dimers. J. Chem. Phys. 1983, 79, 4694–4703. [Google Scholar] [CrossRef]
- Skinner, J.L.; Trommsdorff, H.P. Proton transfer in benzoic acid crystals: A chemical spin–boson problem. Theoretical analysis of nuclear magnetic resonance, neutron scattering, and optical experiments. J. Chem. Phys. 1988, 89, 897–907. [Google Scholar] [CrossRef]
- Horsewill, A.J.; Aibout, A. The dynamics of hydrogen atoms in the hydrogen bonds of carboxylic acid dimers. J. Phys. Condens. Matter 1989, 1, 9609–9622. [Google Scholar] [CrossRef]
- Horsewill, A.J.; Ikram, A.; Tomsah, I.B.I. Hydrogen bond dynamics in tetrafluoroterephthalic acid studied by NMR and INS. Mol. Phys. 1995, 84, 1257–1272. [Google Scholar] [CrossRef]
- Brougham, D.F.; Horsewill, A.J.; Ikram, A.; Ibberson, R.M.; McDonald, P.J.; Pinter-Krainer, M. The correlation between hydrogen bond tunneling dynamics and the structure of benzoic acid dimers. J. Chem. Phys. 1996, 105, 979–982. [Google Scholar] [CrossRef]
- Brougham, D.F.; Horsewill, A.J.; Jenkinson, R.I. Proton transfer dynamics in the hydrogen bond: A direct measurement of the incoherent tunnelling rate by NMR and the quantum-to-classical transition. Chem. Phys. Lett. 1997, 272, 69–74. [Google Scholar] [CrossRef]
- Neumann, M.; Brougham, F.; McGloin, C.J.; Johnson, M.R.; Horsewill, A.J.; Trommsdorff, H.P. Proton tunneling in benzoic acid crystals at intermediate temperatures: Nuclear magnetic resonance and neutron scattering studies. J. Chem. Phys. 1998, 109, 7300–7311. [Google Scholar] [CrossRef]
- Horsewill, A.J.; McGloin, C.J.; Trommsdorff, H.P.; Johnson, M.R. Proton tunnelling in the hydrogen bonds of halogen-substituted derivatives of benzoic acid studied by NMR relaxometry: The case of large energy asymmetry. Chem. Phys. 2003, 291, 41–52. [Google Scholar] [CrossRef]
- Demkin, A.G.; Kolesov, B.A. Tautomeric hydrogen bond in ibuprofen crystals. Phys. Chem. A 2019, 123, 5537–5541. [Google Scholar] [CrossRef]
- Pritchina, E.A.; Kolesov, B.A. Raman spectra of terephthalic acid crystals in the temperature range 5 K–300 K. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 202, 319–323. [Google Scholar] [CrossRef]
- Fischer, P.; Zolliker, P.; Meier, B.H.; Ernst, R.R.; Hewat, A.W.; Jorgensen, J.D.; Rotella, F.J. Structure and Dynamics of Terephthalic Acid from 2 to 300 K. J. Solid State Chem. 1986, 61, 109–125. [Google Scholar] [CrossRef]
- Wilson, C.C.; Shankland, N.; Florence, A.J. Direct determination of the temperature dependence of proton transfer in the benzoic acid dimer by single crystal neutron diffraction. Chem. Phys. Lett. 1996, 253, 103–107. [Google Scholar] [CrossRef]
- Ostrowska, K.; Kropidłowska, M.; Katrusiak, A. High-Pressure Crystallization and Structural Transformations in Compressed R,S-Ibuprofen. Cryst. Growth Des. 2015, 15, 1512–1517. [Google Scholar] [CrossRef]
- Kolesov, B.A. How the vibrational frequency varies with temperature. J. Raman Spectrosc. 2017, 48, 323–326. [Google Scholar] [CrossRef]
- Kolesov, B.A. Proton Delocalization and Tunneling in Terephthalic Acid: Raman Spectroscopic Study. J. Phys. Chem. Solids 2020, 138, 1092888. [Google Scholar] [CrossRef]
- Nakamoto, K.; Margoshes, M.; Rundle, R.E. Stretching Frequencies as a Function of Distances in Hydrogen Bonds. J. Am. Chem. Soc. 1955, 77, 6480–6486. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for Intramolecular N-H···O Resonance-Assisted Hydrogen Bonding in β-Enaminones and Related Heterodienes. A Combined Crystal-Structural, IR and NMR Spectroscopic, and Quantum-Mechanical Investigation. J. Am. Chem. Soc. 2000, 122, 10405–10417. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic Basis of Improper Hydrogen Bonding: A Subtle Balance of Hyperconjugation and Rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef]
- Bent, H.A. An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef] [Green Version]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolesov, B.A. Hydrogen Bonds: Raman Spectroscopic Study. Int. J. Mol. Sci. 2021, 22, 5380. https://doi.org/10.3390/ijms22105380
Kolesov BA. Hydrogen Bonds: Raman Spectroscopic Study. International Journal of Molecular Sciences. 2021; 22(10):5380. https://doi.org/10.3390/ijms22105380
Chicago/Turabian StyleKolesov, Boris A. 2021. "Hydrogen Bonds: Raman Spectroscopic Study" International Journal of Molecular Sciences 22, no. 10: 5380. https://doi.org/10.3390/ijms22105380
APA StyleKolesov, B. A. (2021). Hydrogen Bonds: Raman Spectroscopic Study. International Journal of Molecular Sciences, 22(10), 5380. https://doi.org/10.3390/ijms22105380