
Regulation and Role of Transcription Factors in Osteogenesis


Abstract
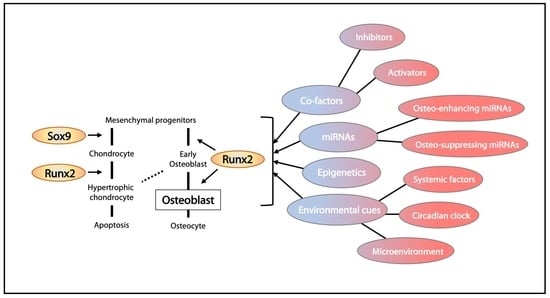
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. An Exciting Era of Bone Biology
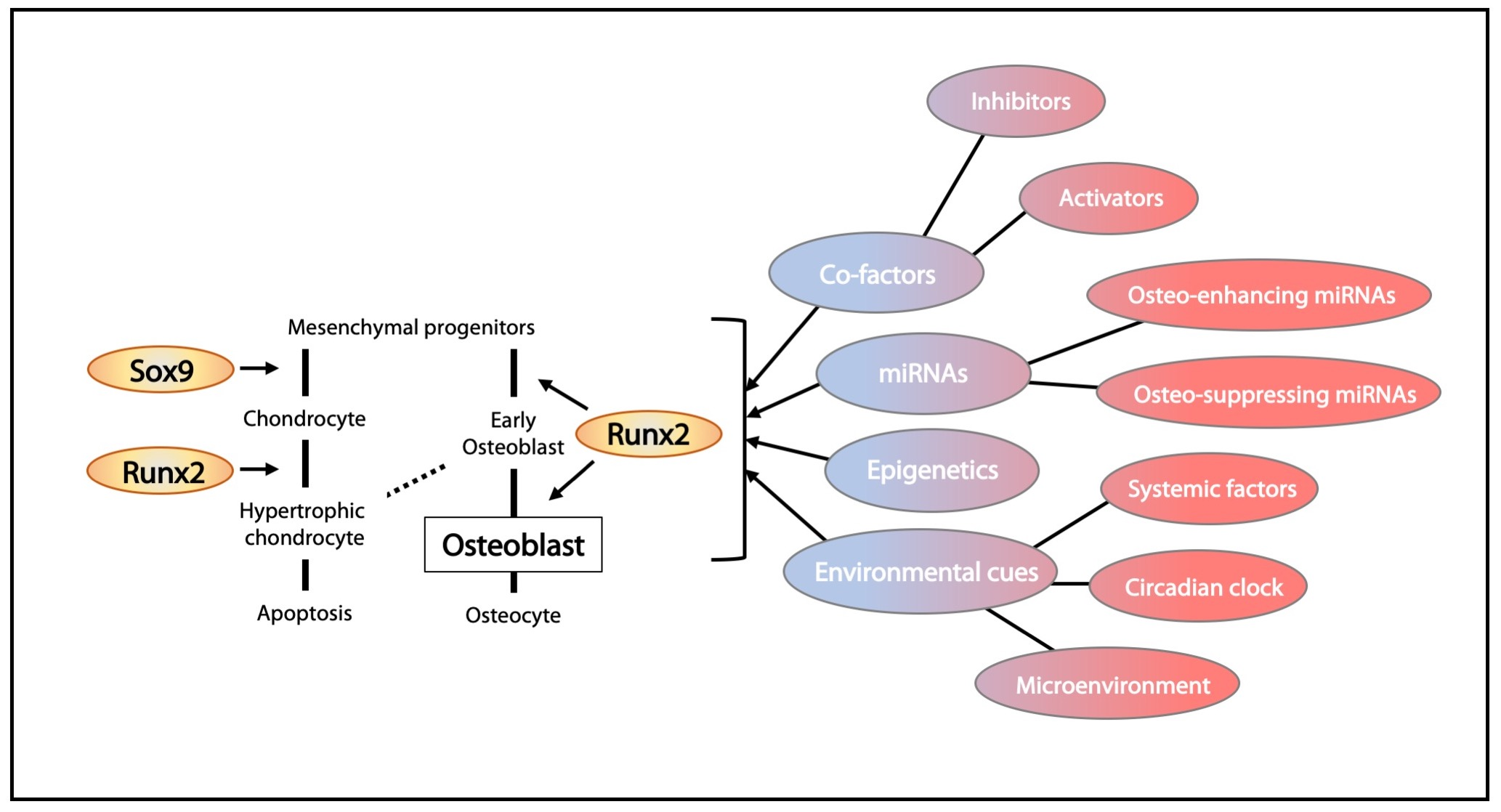
2. Bone Formation in Embryogenesis
2.1. Neural Crest-Derived Osteochondroprogenitors
2.2. Mesoderm-Derived Osteochondroprogenitors
2.3. Endochondral Ossification—Cartilage to Bone Conversion
2.4. Transition of Hypertrophic Chondrocytes to Osteoblasts
2.5. Progenitor Cells for the Continued Growth of Long Bones
2.6. Appositional Bone Growth
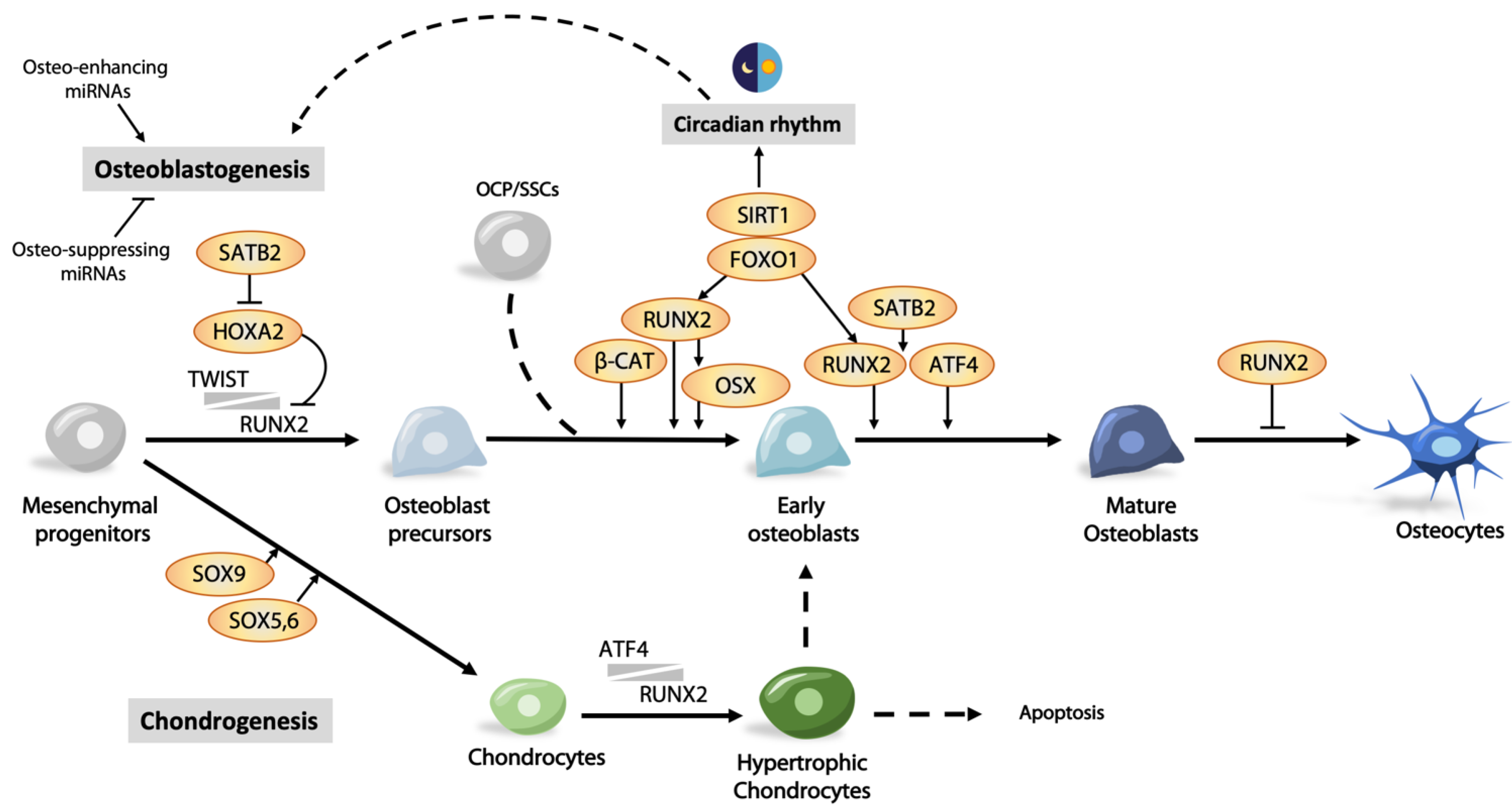
3. Transcriptional Regulation in Osteogenesis
3.1. Runx2 and Its Regulation
3.2. Osx/Sp7 as a Downstream Target of RUNX2
3.3. Other Transcription Factors Regulating Osteoblast Differentiation
3.4. Epigenetic Control of Osteoblast Differentiation
3.4.1. miRNA in Osteoblast Differentiation
3.4.2. DNA and Histone Modifications in Osteoblast Differentiation
3.4.3. Modulating Epigenetic Regulators as Therapy for Bone Disorders
3.5. Regulation of Osteoblast Survival and Death
4. Environmental Cues Regulating Osteogenesis
4.1. Hormonal Control of Osteoblast Differentiation
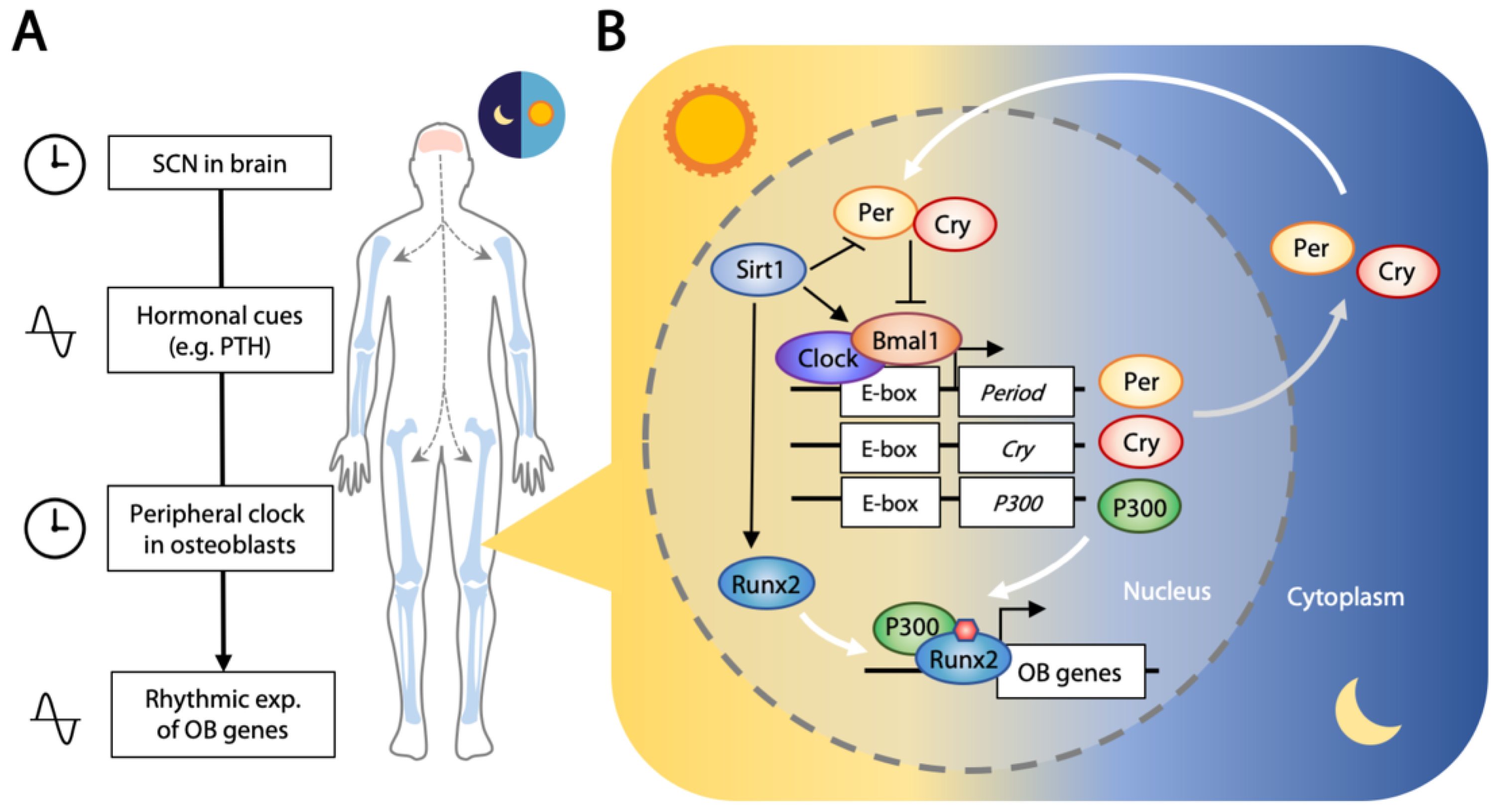
4.2. Circadian Clock Regulates Osteogenesis
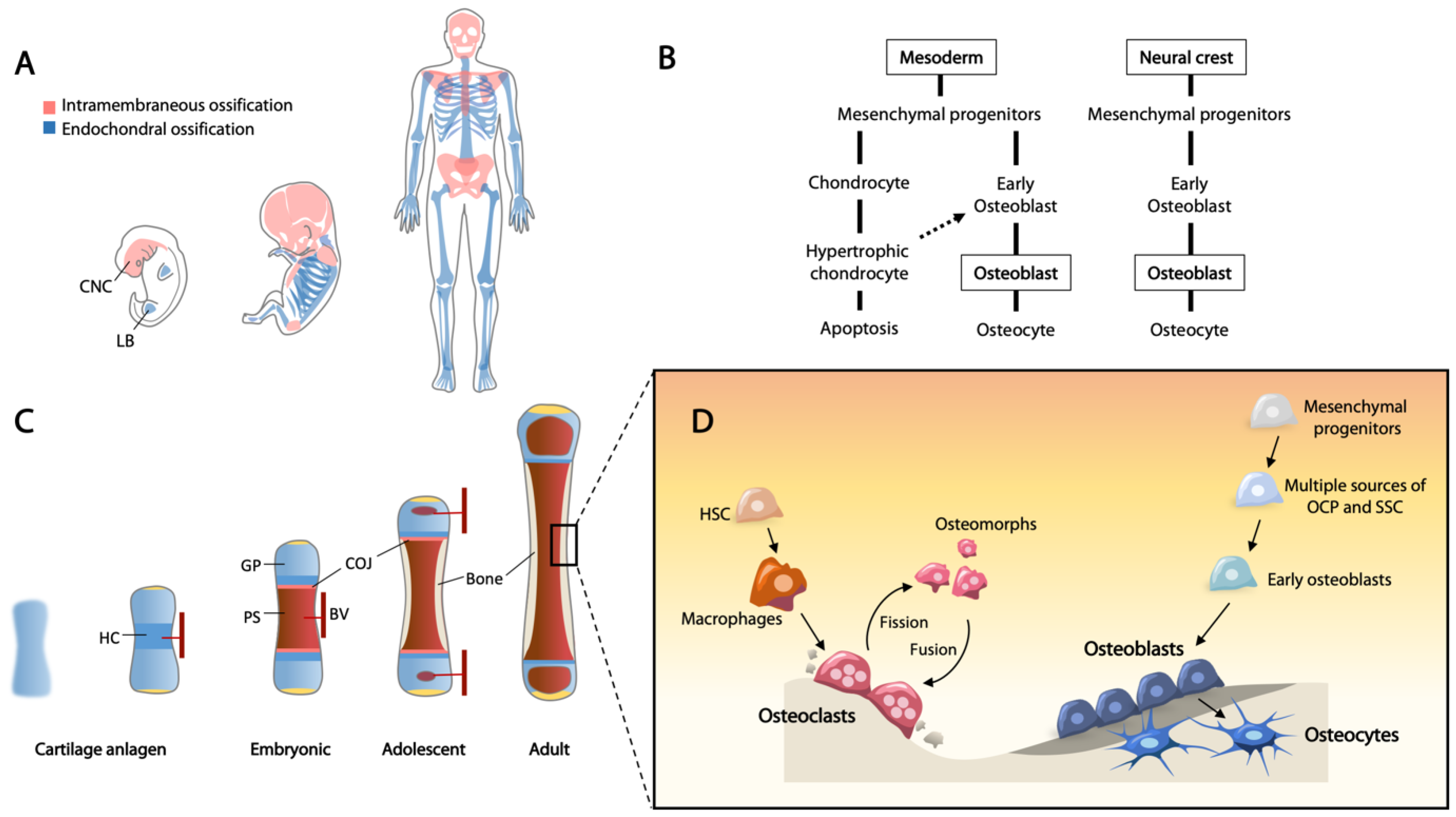
4.3. Molecular Regulation at Sites of Bone Remodeling
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADSC | Adipose tissue-derived stromal cell |
| Cre | Cre recombinase |
| EMT | Epithelial to mesenchymal transition |
| eSSPC | Embryonic skeletal stem/progenitor cell |
| miRNA | microRNA |
| MSC | Mesenchymal stem cell |
| OCP | Osteochondroprogenitor |
| OSE | Osteoblast-specific cis-acting element |
| PTH | Parathyroid hormone |
| SCN | Suprachiasmatic nucleus |
| SSC | Skeletal stem cells |
| UTR | Untranslated regions |
References
- Doro, D.; Liu, A.; Grigoriadis, A.E.; Liu, K.J. The Osteogenic Potential of the Neural Crest Lineage May Contribute to Craniosynostosis. Mol. Syndromol. 2019, 10, 48–57. [Google Scholar] [CrossRef]
- Quarto, N.; Wan, D.C.; Kwan, M.D.; Panetta, N.J.; Li, S.; Longaker, M.T. Origin matters: Differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones. J. Bone Miner. Res. 2010, 25, 1680–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.; Maxson, R.E. Recent advances in craniofacial morphogenesis. Dev. Dyn. 2006, 235, 2353–2375. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Yan, J.; Wang, J.; Zhao, L.; Xin, Q.; Zeng, Y.; Sun, Y.; Zhang, H.; Bai, Z.; Li, Z.; et al. Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.D.; Jayasena, C.S.; Nie, S.; Bronner, M.E. Neural crest specification: Tissues, signals, and transcription factors. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 52–68. [Google Scholar] [CrossRef]
- Roybal, P.G.; Wu, N.L.; Sun, J.; Ting, M.C.; Schafer, C.A.; Maxson, R.E. Inactivation of Msx1 and Msx2 in neural crest reveals an unexpected role in suppressing heterotopic bone formation in the head. Dev. Biol. 2010, 343, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Bildsoe, H.; Loebel, D.A.; Jones, V.J.; Chen, Y.T.; Behringer, R.R.; Tam, P.P. Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev. Biol. 2009, 331, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Chen, G.; Tian, F.; Liu, H.X. Contribution of cranial neural crest cells to mouse skull development. Int. J. Dev. Biol. 2017, 61, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Kamalakar, A.; McKinney, J.M.; Salinas Duron, D.; Amanso, A.M.; Ballestas, S.A.; Drissi, H.; Willett, N.J.; Bhattaram, P.; García, A.J.; Wood, L.B.; et al. JAGGED1 stimulates cranial neural crest cell osteoblast commitment pathways and bone regeneration independent of canonical NOTCH signaling. Bone 2021, 143, 115657. [Google Scholar] [CrossRef]
- Joung, Y.H.; Lim, E.J.; Darvin, P.; Chung, S.C.; Jang, J.W.; Do Park, K.; Lee, H.K.; Kim, H.S.; Park, T.; Yang, Y.M. MSM enhances GH signaling via the Jak2/STAT5b pathway in osteoblast-like cells and osteoblast differentiation through the activation of STAT5b in MSCs. PLoS ONE 2012, 7, e47477. [Google Scholar] [CrossRef]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Hargrave, M.R.; Christiansen, J.; Cooper, L.; Kun, J.; Evans, T.; Gangadharan, U.; Greenfield, A.; Koopman, P. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat. Genet. 1995, 9, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Deng, J.M.; Zhang, Z.; Behringer, R.R.; de Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.W.; Dominguez-Steglich, M.A.; Guioli, S.; Kwok, C.; Weller, P.A.; Stevanovic, M.; Weissenbach, J.; Mansour, S.; Young, I.D.; Goodfellow, P.N.; et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 1994, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E.; et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 1994, 79, 1111–1120. [Google Scholar] [CrossRef]
- Kwok, C.; Weller, P.A.; Guioli, S.; Foster, J.W.; Mansour, S.; Zuffardi, O.; Punnett, H.H.; Dominguez-Steglich, M.A.; Brook, J.D.; Young, I.D.; et al. Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sex reversal. Am. J. Hum. Genet. 1995, 57, 1028–1036. [Google Scholar]
- Wirth, J.; Wagner, T.; Meyer, J.; Pfeiffer, R.A.; Tietze, H.U.; Schempp, W.; Scherer, G. Translocation breakpoints in three patients with campomelic dysplasia and autosomal sex reversal map more than 130 kb from SOX9. Hum. Genet. 1996, 97, 186–193. [Google Scholar] [CrossRef]
- Ikeda, T.; Kawaguchi, H.; Kamekura, S.; Ogata, N.; Mori, Y.; Nakamura, K.; Ikegawa, S.; Chung, U.I. Distinct roles of Sox5, Sox6, and Sox9 in different stages of chondrogenic differentiation. J. Bone Miner. Metab. 2005, 23, 337–340. [Google Scholar] [CrossRef]
- Akiyama, H.; Kim, J.E.; Nakashima, K.; Balmes, G.; Iwai, N.; Deng, J.M.; Zhang, Z.; Martin, J.F.; Behringer, R.R.; Nakamura, T.; et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 2005, 102, 14665–14670. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Zheng, Q.; Engin, F.; Munivez, E.; Chen, Y.; Sebald, E.; Krakow, D.; Lee, B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19004–19009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topol, L.; Chen, W.; Song, H.; Day, T.F.; Yang, Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J. Biol. Chem. 2009, 284, 3323–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dy, P.; Wang, W.; Bhattaram, P.; Wang, Q.; Wang, L.; Ballock, R.T.; Lefebvre, V. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell 2012, 22, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Lian, N.; Li, L.; Moss, H.E.; Perrien, D.S.; Elefteriou, F.; Yang, X. Atf4 regulates chondrocyte proliferation and differentiation during endochondral ossification by activating Ihh transcription. Development 2009, 136, 4143–4153. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Tan, Z.; Niu, B.; Tsang, K.Y.; Tai, A.; Chan, W.C.W.; Lo, R.L.K.; Leung, K.K.H.; Dung, N.W.F.; Itoh, N.; et al. Inhibiting the integrated stress response pathway prevents aberrant chondrocyte differentiation thereby alleviating chondrodysplasia. eLife 2018, 7, e37673. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.M.; Pang, M.K.; Zhou, S.; Cowan, S.K.; Kong, R.Y.; Pfordte, T.; Olsen, B.R.; Sillence, D.O.; Tam, P.P.; Cheah, K.S. Abnormal compartmentalization of cartilage matrix components in mice lacking collagen X: Implications for function. J. Cell Biol. 1997, 136, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Karsenty, G.; Wagner, E.F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2002, 2, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Gerber, H.P.; Hillan, K.J.; Ryan, A.M.; Kowalski, J.; Keller, G.A.; Rangell, L.; Wright, B.D.; Radtke, F.; Aguet, M.; Ferrara, N. VEGF is required for growth and survival in neonatal mice. Development 1999, 126, 1149–1159. [Google Scholar] [CrossRef]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [Green Version]
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell. Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.; Ono, W.; Nagasawa, T.; Kronenberg, H.M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 2014, 16, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhu, L.; Hou, N.; Lan, Y.; Wu, X.M.; Zhou, B.; Teng, Y.; Yang, X. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014, 24, 1266–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Gebhardt, M.; Golovchenko, S.; Perez-Branguli, F.; Hattori, T.; Hartmann, C.; Zhou, X.; deCrombrugghe, B.; Stock, M.; Schneider, H.; et al. Dual pathways to endochondral osteoblasts: A novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 2015, 4, 608–621. [Google Scholar] [CrossRef] [Green Version]
- Tsang, K.Y.; Chan, D.; Cheah, K.S. Fate of growth plate hypertrophic chondrocytes: Death or lineage extension? Dev. Growth Differ. 2015, 57, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Mizuhashi, K.; Ono, W.; Matsushita, Y.; Sakagami, N.; Takahashi, A.; Saunders, T.L.; Nagasawa, T.; Kronenberg, H.M.; Ono, N. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 2018, 563, 254–258. [Google Scholar] [CrossRef]
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014, 10, e1004820. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Kong, M.; Wen, S.; Tsang, K.Y.; Niu, B.; Hartmann, C.; Chan, D.; Hui, C.C.; Cheah, K.S.E. IRX3 and IRX5 Inhibit Adipogenic Differentiation of Hypertrophic Chondrocytes and Promote Osteogenesis. J. Bone Miner. Res. 2020, 35, 2444–2457. [Google Scholar] [CrossRef]
- Ding, M.; Lu, Y.; Abbassi, S.; Li, F.; Li, X.; Song, Y.; Geoffroy, V.; Im, H.J.; Zheng, Q. Targeting Runx2 expression in hypertrophic chondrocytes impairs endochondral ossification during early skeletal development. J. Cell Physiol. 2012, 227, 3446–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, P.; Robey, P.G. Skeletal stem cells. Development 2015, 142, 1023–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the Human Skeletal Stem Cell. Cell 2018, 175, 43–56.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineault, K.M.; Song, J.Y.; Kozloff, K.M.; Lucas, D.; Wellik, D.M. Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat. Commun. 2019, 10, 3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef] [Green Version]
- Carlone, D.L.; Riba-Wolman, R.D.; Deary, L.T.; Tovaglieri, A.; Jiang, L.; Ambruzs, D.M.; Mead, B.E.; Shah, M.S.; Lengner, C.J.; Jaenisch, R.; et al. Telomerase expression marks transitional growth-associated skeletal progenitor/stem cells. Stem Cells 2021, 39, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.R.; Burr, D.B. (Eds.) Bone Growth, Modeling, and Remodeling. In Basic and Applied Bone Biology, 2nd ed.; Elsevier, Inc.: Amsterdam, The Netherlands, 2019; pp. 85–100. [Google Scholar]
- Weaver, C.M.; Peacock, M. Skeletal Changes Across the Life Span. In Basic and Applied Bone Biology, 2nd ed.; Burr, D., Allen, M.R., Eds.; Elsevier, Inc.: Amsterdam, The Netherlands, 2019; pp. 189–202. [Google Scholar]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.P.; Spater, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Liu, M.; Ono, N.; Bringhurst, F.R.; Kronenberg, H.M.; Guo, J. Loss of wnt/beta-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J. Bone Miner. Res. 2012, 27, 2344–2358. [Google Scholar] [CrossRef] [Green Version]
- Houben, A.; Kostanova-Poliakova, D.; Weissenbock, M.; Graf, J.; Teufel, S.; von der Mark, K.; Hartmann, C. beta-catenin activity in late hypertrophic chondrocytes locally orchestrates osteoblastogenesis and osteoclastogenesis. Development 2016, 143, 3826–3838. [Google Scholar] [CrossRef] [Green Version]
- Compton, J.T.; Lee, F.Y. A review of osteocyte function and the emerging importance of sclerostin. J. Bone Jt. Surg. Am. 2014, 96, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bezooijen, R.L.; Roelen, B.A.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; ten Dijke, P.; Lowik, C.W. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 2004, 199, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef]
- Lee, B.; Thirunavukkarasu, K.; Zhou, L.; Pastore, L.; Baldini, A.; Hecht, J.; Geoffroy, V.; Ducy, P.; Karsenty, G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat. Genet. 1997, 16, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Signaling networks in RUNX2-dependent bone development. J. Cell. Biochem. 2010. [Google Scholar] [CrossRef]
- Ducy, P.; Karsenty, G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol. Cell. Biol. 1995, 15, 1858–1869. [Google Scholar] [CrossRef] [Green Version]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, C.A.; Furuichi, T.; Fujita, T.; Fukuyama, R.; Kanatani, N.; Kobayashi, S.; Satake, M.; Takada, K.; Komori, T. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat. Genet. 2002, 32, 633–638. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.B.; Benkusky, N.A.; Pike, J.W. The RUNX2 cistrome in osteoblasts: Characterization, down-regulation following differentiation, and relationship to gene expression. J. Biol. Chem. 2014, 289, 16016–16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Whitfield, T.W.; Gordon, J.A.; Dobson, J.R.; Tai, P.W.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol. 2014, 15, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Toyosawa, S.; Furuichi, T.; Kanatani, N.; Yoshida, C.; Liu, Y.; Himeno, M.; Narai, S.; Yamaguchi, A.; Komori, T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J. Cell Biol. 2001, 155, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, A.; Afzal, F.; Bae, J.S.; Gutierrez, S.; Zaidi, K.; Pratap, J.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs. 2009, 189, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wu, A.; Li, P.; Li, G.; Qin, L.; Song, H.; Mak, K.K. Yap1 Regulates Multiple Steps of Chondrocyte Differentiation during Skeletal Development and Bone Repair. Cell Rep. 2016, 14, 2224–2237. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Lee, H.W.; Lee, J.W.; Kim, J.B. Hes1 stimulates transcriptional activity of Runx2 by increasing protein stabilization during osteoblast differentiation. Biochem. Biophys. Res. Commun. 2008, 367, 97–102. [Google Scholar] [CrossRef]
- Funato, N.; Chapman, S.L.; McKee, M.D.; Funato, H.; Morris, J.A.; Shelton, J.M.; Richardson, J.A.; Yanagisawa, H. Hand2 controls osteoblast differentiation in the branchial arch by inhibiting DNA binding of Runx2. Development 2009, 136, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Shimazu, J.; Makinistoglu, M.P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Lezaki, T.; Pessin, J.E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161, 1576–1591. [Google Scholar] [CrossRef] [Green Version]
- Bialek, P.; Kern, B.; Yang, X.; Schrock, M.; Sosic, D.; Hong, N.; Wu, H.; Yu, K.; Ornitz, D.M.; Olson, E.N.; et al. A twist code determines the onset of osteoblast differentiation. Dev. Cell 2004, 6, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.Q.; Javed, A.; Morasso, M.I.; Karlin, J.; Montecino, M.; van Wijnen, A.J.; Stein, G.S.; Stein, J.L.; Lian, J.B. Dlx3 transcriptional regulation of osteoblast differentiation: Temporal recruitment of Msx2, Dlx3, and Dlx5 homeodomain proteins to chromatin of the osteocalcin gene. Mol. Cell. Biol. 2004, 24, 9248–9261. [Google Scholar] [CrossRef] [Green Version]
- Satokata, I.; Ma, L.; Ohshima, H.; Bei, M.; Woo, I.; Nishizawa, K.; Maeda, T.; Takano, Y.; Uchiyama, M.; Heaney, S.; et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 2000, 24, 391–395. [Google Scholar] [CrossRef]
- Zhang, J.; Tu, Q.; Grosschedl, R.; Kim, M.S.; Griffin, T.; Drissi, H.; Yang, P.; Chen, J. Roles of SATB2 in osteogenic differentiation and bone regeneration. Tissue Eng. Part A 2011, 17, 1767–1776. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Baek, W.Y.; Kim, J.E. Transcriptional regulation of bone formation. Front. Biosci. 2011, 3, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Baek, W.Y.; Lee, M.A.; Jung, J.W.; Kim, S.Y.; Akiyama, H.; de Crombrugghe, B.; Kim, J.E. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J. Bone Miner. Res. 2009, 24, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, Z.; Feng, J.Q.; Dusevich, V.M.; Sinha, K.; Zhang, H.; Darnay, B.G.; de Crombrugghe, B. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc. Natl. Acad. Sci. USA 2010, 107, 12919–12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducy, P.; Starbuck, M.; Priemel, M.; Shen, J.; Pinero, G.; Geoffroy, V.; Amling, M.; Karsenty, G. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999, 13, 1025–1036. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Kwon, T.G.; Park, H.S.; Wozney, J.M.; Ryoo, H.M. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochem. Biophys. Res. Commun. 2003, 309, 689–694. [Google Scholar] [CrossRef]
- Koga, T.; Matsui, Y.; Asagiri, M.; Kodama, T.; de Crombrugghe, B.; Nakashima, K.; Takayanagi, H. NFAT and Osterix cooperatively regulate bone formation. Nat. Med. 2005, 11, 880–885. [Google Scholar] [CrossRef]
- Zhang, C.; Cho, K.; Huang, Y.; Lyons, J.P.; Zhou, X.; Sinha, K.; McCrea, P.D.; de Crombrugghe, B. Inhibition of Wnt signaling by the osteoblast-specific transcription factor Osterix. Proc. Natl. Acad. Sci. USA 2008, 105, 6936–6941. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M. Unraveling the role of FoxOs in bone—Insights from mouse models. Bone 2011, 49, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Gong, Y.; Xu, L.; Zhou, M.; Li, J.; Song, J. Bidirectional regulation of osteogenic differentiation by the FOXO subfamily of Forkhead transcription factors in mammalian MSCs. Cell Prolif. 2019, 52, e12540. [Google Scholar] [CrossRef] [Green Version]
- Rached, M.T.; Kode, A.; Xu, L.; Yoshikawa, Y.; Paik, J.H.; Depinho, R.A.; Kousteni, S. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010, 11, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Makowski, A.J.; Uppuganti, S.; Wadeer, S.A.; Whitehead, J.M.; Rowland, B.J.; Granke, M.; Mahadevan-Jansen, A.; Yang, X.; Nyman, J.S. The loss of activating transcription factor 4 (ATF4) reduces bone toughness and fracture toughness. Bone 2014, 62, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dobreva, G.; Chahrour, M.; Dautzenberg, M.; Chirivella, L.; Kanzler, B.; Fariñas, I.; Karsenty, G.; Grosschedl, R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 2006, 125, 971–986. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H.C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T.M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology: Implication for Coffin-Lowry Syndrome. Cell 2004, 117, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Franceschi, R.T.; Luo, M.; Fan, J.; Jiang, D.; Cao, H.; Kwon, T.G.; Lai, Y.; Zhang, J.; Patrene, K.; et al. Critical role of activating transcription factor 4 in the anabolic actions of parathyroid hormone in bone. PLoS ONE 2009, 4, e7583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elefteriou, F.; Benson, M.D.; Sowa, H.; Starbuck, M.; Liu, X.; Ron, D.; Parada, L.F.; Karsenty, G. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006, 4, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Grotewold, L.; Ruther, U. The Fused toes (Ft) mouse mutation causes anteroposterior and dorsoventral polydactyly. Dev. Biol. 2002, 251, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, T.; Ausmeier, K.; Dildrop, R.; Ruther, U. The mouse Fused toes (Ft) mutation is the result of a 1.6-Mb deletion including the entire Iroquois B gene cluster. Mamm. Genome 2002, 13, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Hamamy, H.A.; Masri, A.T.; Al-Hadidy, A.M.; Ajlouni, K.M. Consanguinity and genetic disorders. Profile from Jordan. Saudi Med. J. 2007, 28, 1015–1017. [Google Scholar] [PubMed]
- Hamamy, H.A.; Teebi, A.S.; Oudjhane, K.; Shegem, N.N.; Ajlouni, K.M. Severe hypertelorism, midface prominence, prominent/simple ears, severe myopia, borderline intelligence, and bone fragility in two brothers: New syndrome? Am. J. Med. Genet. Part A 2007, 143, 229–234. [Google Scholar] [CrossRef]
- Bonnard, C.; Strobl, A.C.; Shboul, M.; Lee, H.; Merriman, B.; Nelson, S.F.; Ababneh, O.H.; Uz, E.; Guran, T.; Kayserili, H.; et al. Mutations in IRX5 impair craniofacial development and germ cell migration via SDF1. Nat. Genet. 2012, 44, 709–713. [Google Scholar] [CrossRef]
- Chang, C.F.; Li, L.H.; Wang, C.H.; Tsai, F.J.; Chen, T.C.; Wu, J.Y.; Chen, Y.T.; Tsai, A.C. Identification of a submicroscopic 3.2 Mb chromosomal 16q12.2-13 deletion in a child with short stature, mild developmental delay, and craniofacial anomalies, by high-density oligonucleotide array—A recognizable syndrome. Am. J. Med. Genet. Part A 2010, 152, 2365–2371. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Sakuma, R.; Wylie, J.N.; Kim, K.H.; Zhang, S.S.; Hui, C.C.; Bruneau, B.G. Cooperative and antagonistic roles for Irx3 and Irx5 in cardiac morphogenesis and postnatal physiology. Development 2012, 139, 4007–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Sakuma, R.; Vakili, N.A.; Mo, R.; Puviindran, V.; Deimling, S.; Zhang, X.; Hopyan, S.; Hui, C.C. Formation of proximal and anterior limb skeleton requires early function of Irx3 and Irx5 and is negatively regulated by Shh signaling. Dev. Cell 2014, 29, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, C.J.; Gaborit, N.; Lwin, W.; Barruet, E.; Ho, S.; Bonnard, C.; Hamamy, H.; Shboul, M.; Reversade, B.; Kayserili, H.; et al. Loss of Iroquois homeobox transcription factors 3 and 5 in osteoblasts disrupts cranial mineralization. Bone Rep. 2016, 5, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justesen, J.; Stenderup, K.; Ebbesen, E.N.; Mosekilde, L.; Steiniche, T.; Kassem, M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2001, 2, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Tu, Q.; Meng, S.; Zhang, L.; Yu, L.; Song, J.; Hu, Y.; Sui, L.; Zhang, J.; Dard, M.; et al. Runx2/DICER/miRNA Pathway in Regulating Osteogenesis. J. Cell Physiol. 2017, 232, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Baumgart, M.; Groth, M.; Wittmann, J.; Jäck, H.M.; Platzer, M.; Tuckermann, J.P.; Baschant, U. Dicer ablation in osteoblasts by Runx2 driven cre-loxP recombination affects bone integrity, but not glucocorticoid-induced suppression of bone formation. Sci. Rep. 2016, 6, 32112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, T.; Hussain, S.; Mudhasani, R.; Parulkar, I.; Colby, J.L.; Frederick, D.; Kream, B.E.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Dicer inactivation in osteoprogenitor cells compromises fetal survival and bone formation, while excision in differentiated osteoblasts increases bone mass in the adult mouse. Dev. Biol. 2010, 340, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, A.P.; McAlinden, A. The role of microRNAs in bone development. Bone 2021, 143, 115760. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Hou, C.; Meng, F.; Zhao, X.; Huang, G.; Chen, W.; Fu, M.; Liao, W. MiR-455-3p regulates early chondrogenic differentiation via inhibiting Runx2. FEBS Lett. 2015, 589, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, B.; Vishal, M.; Shreya, S.; Malavika, D.; Rajpriya, V.; He, Z.; Partridge, N.C.; Selvamurugan, N. Parathyroid hormone-stimulation of Runx2 during osteoblast differentiation via the regulation of lnc-SUPT3H-1:16 (RUNX2-AS1:32) and miR-6797-5p. Biochimie 2019, 158, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhong, L.; Yuan, T.; Chen, S.; Zhou, Y.; An, L.; Guo, Y.; Fan, M.; Li, Y.; Sun, Y.; et al. MicroRNA-155 inhibits the osteogenic differentiation of mesenchymal stem cells induced by BMP9 via downregulation of BMP signaling pathway. Int. J. Mol. Med. 2018, 41, 3379–3393. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Jensen, E.D.; Westendorf, J.J. Runx2: A master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res. C Embryo Today 2005, 75, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, Z.; Hou, T.; Li, Z.; Huang, K.; Gong, J.; Zhou, W.; Tang, K.; Xu, J.; Dong, S. MiR-125b Regulates the Osteogenic Differentiation of Human Mesenchymal Stem Cells by Targeting BMPR1b. Cell. Physiol. Biochem. 2017, 41, 530–542. [Google Scholar] [CrossRef]
- Cao, Y.; LV, Q.; LV, C. MicroRNA-153 suppresses the osteogenic differentiation of human mesenchymal stem cells by targeting bone morphogenetic protein receptor type II. Int. J. Mol. Med. 2015, 36, 760–766. [Google Scholar] [CrossRef]
- Zeng, Y.; Qu, X.; Li, H.; Huang, S.; Wang, S.; Xu, Q.; Lin, R.; Han, Q.; Li, J.; Zhao, R.C. MicroRNA-100 regulates osteogenic differentiation of human adipose-derived mesenchymal stem cells by targeting BMPR2. FEBS Lett. 2012, 586, 2375–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyama, K.; Yamada, G.; Yamamoto, T.S.; Takagi, C.; Miyado, K.; Sakai, M.; Ueno, N.; Shibuya, H. A BMP-inducible gene, dlx5, regulates osteoblast differentiation and mesoderm induction. Dev. Biol. 1999, 208, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.Q.; Tare, R.; Lee, S.H.; Mandeville, M.; Weiner, B.; Montecino, M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. HOXA10 controls osteoblastogenesis by directly activating bone regulatory and phenotypic genes. Mol. Cell. Biol. 2007, 27, 3337–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Meng, Y.; Liu, Y.; Chen, Y.; Yang, H.; Chen, D.; Shi, J.; Guo, Y. MicroRNA-320a Regulates the Osteogenic Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells by Targeting HOXA10. Cell. Physiol. Biochem. 2016, 38, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Nozawa, Y.; Akao, Y. MicroRNA-141 and -200a are involved in bone morphogenetic protein-2-induced mouse pre-osteoblast differentiation by targeting distal-less homeobox 5. J. Biol. Chem. 2009, 284, 19272–19279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westendorf, J.J. Transcriptional co-repressors of Runx2. J. Cell. Biochem. 2006, 98, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Liu, W.; Li, H.; Yang, L.; Chen, C.; Xia, Z.Y.; Guo, L.J.; Xie, H.; Zhou, H.D.; Wu, X.P.; et al. A Runx2/miR-3960/miR-2861 regulatory feedback loop during mouse osteoblast differentiation. J. Biol. Chem. 2011, 286, 12328–12339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hassan, M.Q.; Jafferji, M.; Aqeilan, R.I.; Garzon, R.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J. Biol. Chem. 2009, 284, 15676–15684. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Hong, F.; Bao, Q.; Xu, Q.; Duan, R.; Zhu, Z.; Zhang, W.; Ma, C. MicroRNA-145 suppresses osteogenic differentiation of human jaw bone marrow mesenchymal stem cells partially via targeting semaphorin 3A. Connect. Tissue Res. 2020, 61, 577–585. [Google Scholar] [CrossRef]
- Jia, J.; Tian, Q.; Ling, S.; Liu, Y.; Yang, S.; Shao, Z. miR-145 suppresses osteogenic differentiation by targeting Sp7. FEBS Lett. 2013, 587, 3027–3031. [Google Scholar] [CrossRef] [Green Version]
- Bellavia, D.; Salamanna, F.; Raimondi, L.; De Luca, A.; Carina, V.; Costa, V.; Alessandro, R.; Fini, M.; Giavaresi, G. Deregulated miRNAs in osteoporosis: Effects in bone metastasis. Cell. Mol. Life Sci. 2019, 76, 3723–3744. [Google Scholar] [CrossRef]
- Bottani, M.; Banfi, G.; Lombardi, G. The Clinical Potential of Circulating miRNAs as Biomarkers: Present and Future Applications for Diagnosis and Prognosis of Age-Associated Bone Diseases. Biomolecules 2020, 10, 589. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.; Heilmeier, U.; Weilner, S.; Grillari, J. Circulating microRNAs as novel biomarkers for bone diseases—Complex signatures for multifactorial diseases? Mol. Cell. Endocrinol. 2016, 432, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Grillari, J.; Mäkitie, R.E.; Kocijan, R.; Haschka, J.; Vázquez, D.C.; Semmelrock, E.; Hackl, M. Circulating miRNAs in bone health and disease. Bone 2021, 145, 115787. [Google Scholar] [CrossRef]
- Yang, R.; Yu, T.; Kou, X.; Gao, X.; Chen, C.; Liu, D.; Zhou, Y.; Shi, S. Tet1 and Tet2 maintain mesenchymal stem cell homeostasis via demethylation of the P2rX7 promoter. Nat. Commun. 2018, 9, 2143. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Zainabadi, K.; Bell, E.; Sykes, D.B.; Saez, B.; Lotinun, S.; Baron, R.; Scadden, D.; Schipani, E.; Guarente, L. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating β-catenin. EMBO Mol. Med. 2013, 5, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Ghorbaninejad, M.; Khademi-Shirvan, M.; Hosseini, S.; Baghaban Eslaminejad, M. Epidrugs: Novel epigenetic regulators that open a new window for targeting osteoblast differentiation. Stem Cell Res. Ther. 2020, 11, 456. [Google Scholar] [CrossRef]
- Chen, Y.S.; Wu, R.; Yang, X.; Kou, S.; MacDougald, O.A.; Yu, L.; Shi, H.; Xue, B. Inhibiting DNA methylation switches adipogenesis to osteoblastogenesis by activating Wnt10a. Sci. Rep. 2016, 6, 25283. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Lee, Y.M.; Kim, M.J.; Choi, J.Y.; Park, E.K.; Kim, S.Y.; Lee, S.P.; Yang, J.S.; Kim, D.S. Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol. Cells 2006, 22, 182–188. [Google Scholar]
- Cho, H.H.; Park, H.T.; Kim, Y.J.; Bae, Y.C.; Suh, K.T.; Jung, J.S. Induction of osteogenic differentiation of human mesenchymal stem cells by histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 533–542. [Google Scholar] [CrossRef]
- Chen, T.H.; Chen, W.M.; Hsu, K.H.; Kuo, C.D.; Hung, S.C. Sodium butyrate activates ERK to regulate differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2007, 355, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Fu, Y.; Zhang, X.; Dai, L.; Zhu, J.; Bi, Z.; Ao, Y.; Zhou, C. Histone deacetylase inhibitor sodium butyrate promotes the osteogenic differentiation of rat adipose-derived stem cells. Dev. Growth Differ. 2014, 56, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, B.C.; Bilezikian, J.P. Parathyroid hormone: Anabolic and catabolic actions on the skeleton. Curr. Opin. Pharm. 2015, 22, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isogai, Y.; Akatsu, T.; Ishizuya, T.; Yamaguchi, A.; Hori, M.; Takahashi, N.; Suda, T. Parathyroid hormone regulates osteoblast differentiation positively or negatively depending on the differentiation stages. J. Bone Miner. Res. 1996, 11, 1384–1393. [Google Scholar] [CrossRef]
- Bellido, T.; Ali, A.A.; Plotkin, L.I.; Fu, Q.; Gubrij, I.; Roberson, P.K.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 2003, 278, 50259–50272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, Y.; Iwasawa, M.; Akiyama, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Yasui, T.; Matsumoto, T.; Hirose, J.; Nakamura, H.; et al. Anti-apoptotic molecule Bcl-2 regulates the differentiation, activation, and survival of both osteoblasts and osteoclasts. J. Biol. Chem. 2009, 284, 36659–36669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriishi, T.; Kawai, Y.; Komori, H.; Rokutanda, S.; Eguchi, Y.; Tsujimoto, Y.; Asahina, I.; Komori, T. Bcl2 deficiency activates FoxO through Akt inactivation and accelerates osteoblast differentiation. PLoS ONE 2014, 9, e86629. [Google Scholar] [CrossRef]
- Wang, X.; Kua, H.Y.; Hu, Y.; Guo, K.; Zeng, Q.; Wu, Q.; Ng, H.H.; Karsenty, G.; de Crombrugghe, B.; Yeh, J.; et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 2006, 172, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Cell Death in Chondrocytes, Osteoblasts, and Osteocytes. Int. J. Mol. Sci. 2016, 17, 2045. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Liu, W.; Cao, H.; Xiao, G. Molecular mechanosensors in osteocytes. Bone Res. 2020, 8, 23. [Google Scholar] [CrossRef]
- Guasto, A.; Cormier-Daire, V. Signaling Pathways in Bone Development and Their Related Skeletal Dysplasia. Int. J. Mol. Sci. 2021, 22, 4321. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.K.Y.; Chan, A.S.F.; Rubab, A.; Chan, W.C.W.; Chan, D. Extracellular Matrix and Cellular Plasticity in Musculoskeletal Development. Front. Cell Dev. Biol. 2020, 8, 781. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.; Reifenstein, E.C.; Forbes, A.P. Effect of stilbestrol in post-menopausal osteoporosis. Trans. Conf. Metab. Asp. Conval 1946, 14, 99–101. [Google Scholar]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Carboni, G.A.; Guemes, M.; Bailey, S.; Anaya, E.; Corselli, M.; Peault, B.; Krum, S.A. GATA4 regulates estrogen receptor-alpha-mediated osteoblast transcription. Mol. Endocrinol. 2011, 25, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Khalid, A.B.; Slayden, A.V.; Kumpati, J.; Perry, C.D.; Osuna, M.A.L.; Arroyo, S.R.; Miranda-Carboni, G.A.; Krum, S.A. GATA4 Directly Regulates. JBMR Plus 2018, 2, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Griffin, E.A.; Staknis, D.; Weitz, C.J. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science 1999, 286, 768–771. [Google Scholar] [CrossRef]
- Shearman, L.P.; Sriram, S.; Weaver, D.R.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; van der Horst, G.T.; Hastings, M.H.; et al. Interacting molecular loops in the mammalian circadian clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [Green Version]
- Dickmeis, T.; Weger, B.D.; Weger, M. The circadian clock and glucocorticoids--interactions across many time scales. Mol. Cell. Endocrinol. 2013, 380, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.F.; Meng, Q.J. Timing metabolism in cartilage and bone: Links between circadian clocks and tissue homeostasis. J. Endocrinol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Kume, K.; Zylka, M.J.; Sriram, S.; Shearman, L.P.; Weaver, D.R.; Jin, X.; Maywood, E.S.; Hastings, M.H.; Reppert, S.M. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 1999, 98, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Etchegaray, J.P.; Cagampang, F.R.; Loudon, A.S.; Reppert, S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef]
- Gallego, M.; Virshup, D.M. Post-translational modifications regulate the ticking of the circadian clock. Nat. Rev. Mol. Cell Biol. 2007, 8, 139–148. [Google Scholar] [CrossRef]
- Lowrey, P.L.; Takahashi, J.S. Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 2011, 74, 175–230. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Yang, S.F.; Wang, Z.; Tan, J.M.; Xing, S.M.; Chen, D.C.; Xu, S.M.; Yuan, W. PCAF acetylates Runx2 and promotes osteoblast differentiation. J. Bone Miner. Metab. 2013, 31, 381–389. [Google Scholar] [CrossRef]
- Tang, Z.; Xu, T.; Li, Y.; Fei, W.; Yang, G.; Hong, Y. Inhibition of CRY2 by STAT3/miRNA-7-5p Promotes Osteoblast Differentiation through Upregulation of CLOCK/BMAL1/P300 Expression. Mol. Ther. Nucleic Acids 2020, 19, 865–876. [Google Scholar] [CrossRef]
- Feskanich, D.; Hankinson, S.E.; Schernhammer, E.S. Nightshift work and fracture risk: The Nurses’ Health Study. Osteoporos. Int. 2009, 20, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wang, J.; Kim, B.; Lu, C.; Zhang, Z.; Liu, H.; Kang, H.; Sun, Y.; Guan, H.; Fang, Z.; et al. Insights into the Role of Circadian Rhythms in Bone Metabolism: A Promising Intervention Target? Biomed. Res. Int. 2018, 2018, 9156478. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Bruscalupi, G. Circadian Rhythms and Hormonal Homeostasis: Pathophysiological Implications. Biology 2017, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- El-Hajj Fuleihan, G.; Klerman, E.B.; Brown, E.N.; Choe, Y.; Brown, E.M.; Czeisler, C.A. The parathyroid hormone circadian rhythm is truly endogenous—A general clinical research center study. J. Clin. Endocrinol. Metab. 1997, 82, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Patel, M.S.; Bradley, A.; Wagner, E.F.; Karsenty, G. The molecular clock mediates leptin-regulated bone formation. Cell 2005, 122, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanyu, R.; Hayata, T.; Nagao, M.; Saita, Y.; Hemmi, H.; Notomi, T.; Nakamoto, T.; Schipani, E.; Knonenbery, H.; Kaneko, K.; et al. Per-1 is a specific clock gene regulated by parathyroid hormone (PTH) signaling in osteoblasts and is functional for the transcriptional events induced by PTH. J. Cell. Biochem. 2011, 112, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Okubo, N.; Fujiwara, H.; Minami, Y.; Kunimoto, T.; Hosokawa, T.; Umemura, Y.; Inokawa, H.; Asada, M.; Oda, R.; Kubo, T.; et al. Parathyroid hormone resets the cartilage circadian clock of the organ-cultured murine femur. Acta Orthop. 2015, 86, 627–631. [Google Scholar] [CrossRef]
- Zvonic, S.; Ptitsyn, A.A.; Kilroy, G.; Wu, X.; Conrad, S.A.; Scott, L.K.; Guilak, F.; Pelled, G.; Gazit, D.; Gimble, J.M. Circadian oscillation of gene expression in murine calvarial bone. J. Bone Miner. Res. 2007, 22, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Okubo, N.; Minami, Y.; Fujiwara, H.; Umemura, Y.; Tsuchiya, Y.; Shirai, T.; Oda, R.; Inokawa, H.; Kubo, T.; Yagita, K. Prolonged bioluminescence monitoring in mouse ex vivo bone culture revealed persistent circadian rhythms in articular cartilages and growth plates. PLoS ONE 2013, 8, e78306. [Google Scholar] [CrossRef] [Green Version]
- Kunimoto, T.; Okubo, N.; Minami, Y.; Fujiwara, H.; Hosokawa, T.; Asada, M.; Oda, R.; Kubo, T.; Yagita, K. A PTH-responsive circadian clock operates in ex vivo mouse femur fracture healing site. Sci. Rep. 2016, 6, 22409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gafni, Y.; Ptitsyn, A.A.; Zilberman, Y.; Pelled, G.; Gimble, J.M.; Gazit, D. Circadian rhythm of osteocalcin in the maxillomandibular complex. J. Dent. Res. 2009, 88, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nizkorodov, A.; Riemenschneider, K.; Lee, C.S.; Olivares-Navarrete, R.; Schwartz, Z.; Boyan, B.D. Impaired bone formation in Pdia3 deficient mice. PLoS ONE 2014, 9, e112708. [Google Scholar] [CrossRef]
- Yuan, G.; Hua, B.; Yang, Y.; Xu, L.; Cai, T.; Sun, N.; Yan, Z.; Lu, C.; Qian, R. The Circadian Gene Clock Regulates Bone Formation Via PDIA3. J. Bone Miner. Res. 2017, 32, 861–871. [Google Scholar] [CrossRef]
- Samsa, W.E.; Vasanji, A.; Midura, R.J.; Kondratov, R.V. Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone 2016, 84, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dernie, F.; Adeyoju, D. A matter of time: Circadian clocks in osteoarthritis and the potential of chronotherapy. Exp. Gerontol. 2021, 143, 111163. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 2007, 450, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakibaei, M.; Shayan, P.; Busch, F.; Aldinger, C.; Buhrmann, C.; Lueders, C.; Mobasheri, A. Resveratrol mediated modulation of Sirt-1/Runx2 promotes osteogenic differentiation of mesenchymal stem cells: Potential role of Runx2 deacetylation. PLoS ONE 2012, 7, e35712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zainabadi, K.; Liu, C.J.; Guarente, L. SIRT1 is a positive regulator of the master osteoblast transcription factor, RUNX2. PLoS ONE 2017, 12, e0178520. [Google Scholar] [CrossRef] [Green Version]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Khoo, W.H.; Ng, P.Y.; Xiao, Y.; Zamerli, J.; Thatcher, P.; Kyaw, W.; Pathmanandavel, K.; Grootveld, A.K.; Moran, I.; et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 2021, 184, 1940. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. S1) (Suppl. S1), 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Gasser, A.; Bolamperti, S.; Maeda, M.; Matthies, L.; Jähn, K.; Long, C.L.; Schlüter, H.; Kwiatkowski, M.; Saini, V.; et al. TG-interacting factor 1 (Tgif1)-deficiency attenuates bone remodeling and blunts the anabolic response to parathyroid hormone. Nat. Commun. 2019, 10, 1354. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, W.C.W.; Tan, Z.; To, M.K.T.; Chan, D. Regulation and Role of Transcription Factors in Osteogenesis. Int. J. Mol. Sci. 2021, 22, 5445. https://doi.org/10.3390/ijms22115445
Chan WCW, Tan Z, To MKT, Chan D. Regulation and Role of Transcription Factors in Osteogenesis. International Journal of Molecular Sciences. 2021; 22(11):5445. https://doi.org/10.3390/ijms22115445
Chicago/Turabian StyleChan, Wilson Cheuk Wing, Zhijia Tan, Michael Kai Tsun To, and Danny Chan. 2021. "Regulation and Role of Transcription Factors in Osteogenesis" International Journal of Molecular Sciences 22, no. 11: 5445. https://doi.org/10.3390/ijms22115445
APA StyleChan, W. C. W., Tan, Z., To, M. K. T., & Chan, D. (2021). Regulation and Role of Transcription Factors in Osteogenesis. International Journal of Molecular Sciences, 22(11), 5445. https://doi.org/10.3390/ijms22115445