
Zebrafish as a Model for the Study of Lipid-Lowering Drug-Induced Myopathies

 , , ,
, , ,
Abstract
:1. Introduction
2. Pathological Mechanisms Underlying Lipid-Lowering Drug-Induced Myopathies
2.1. Statins
2.2. Fibrates
2.3. Ezetimibe
3. Models for Study of Lipid-Lowering Drug-Induced Myopathies
4. Zebrafish Models for Study of Lipid-Lowering Drug-Induced Myopathies
5. Zebrafish Usefulness in Research Dealing with a Lipid-Lowering Drug Environmental Issue
6. Perspectives
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, D.; Li, A.; Zhou, H.; Wang, X.; Li, P.; Bi, S.; Teng, Y. Uncover the Underlying Mechanism of Drug-Induced Myopathy by Using Systems Biology Approaches. Int. J. Genom. 2017, 2017, 9264034. [Google Scholar] [CrossRef] [Green Version]
- Pasnoor, M.; Barohn, R.J.; Dimachkie, M.M. Toxic Myopathies. Neurol. Clin. 2014, 32, 647–670. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C. Toxic and Drug-Induced Myopathies. J. Neurol. Neurosurg. Psychiatry 2009, 80, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Alcantara, R.; Popli, T.; Mahajan, S.; Tariq, U.; Dusaj, R.S.; Malik, A.H. Myopathy Associated with Statins and SGLT2—A Review of Literature. Curr. Probl. Cardiol. 2021, 46, 100765. [Google Scholar] [CrossRef] [PubMed]
- Zodda, D.; Giammona, R.; Schifilliti, S. Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs. Pharmacy 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, L.E.; Stoekenbroek, R.M.; Kastelein, J.J.P.; Holleboom, A.G. Moving Targets: Recent Advances in Lipid-Lowering Therapies. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Doughty, C.T.; Amato, A.A. Toxic Myopathies. Contin. Lifelong Learn. Neurol. 2019, 25, 1712–1731. [Google Scholar] [CrossRef]
- Bouitbir, J.; Sanvee, G.M.; Panajatovic, M.V.; Singh, F.; Krähenbühl, S. Mechanisms of Statin-Associated Skeletal Muscle-Associated Symptoms. Pharmacol. Res. 2020, 154, 104201. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein Sensors for Membrane Sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M.; et al. Intensive versus Moderate Lipid Lowering with Statins after Acute Coronary Syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Kavalipati, N.; Shah, J.; Ramakrishan, A.; Vasnawala, H. Pleiotropic Effects of Statins. Indian J. Endocrinol. Metab. 2015, 19, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Verdoodt, A.; Honore, P.M.; Jacobs, R.; De Waele, E.; Van Gorp, V.; De Regt, J.; Spapen, H.D. Do Statins Induce or Protect from Acute Kidney Injury and Chronic Kidney Disease: An Update Review in 2018. J. Transl. Int. Med. 2018, 6, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmeijer, K.; Dekkers, O.M.; de Fijter, J.W.; Dekker, F.W.; Hoogeveen, E.K. Effect of Different Types of Statins on Kidney Function Decline and Proteinuria: A Network Meta-Analysis. Sci. Rep. 2019, 9, 16632. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.-M.; Wu, S.-G.; Chen, F.; Wu, Q.; Wu, C.-M.; Kang, C.-M.; He, X.; Zhang, R.-Y.; Lu, Z.-F.; Li, X.-H.; et al. Atorvastatin Inhibits Pyroptosis through the LncRNA NEXN-AS1/NEXN Pathway in Human Vascular Endothelial Cells. Atherosclerosis 2020, 293, 26–34. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Morita, I.; Sato, I.; Ma, L.; Murota, S.-I. Enhancement of Membrane Fluidity in Cholesterol-Poor Endothelial Cells Pre-Treated with Simvastatin. Endothelium 1997, 5, 107–113. [Google Scholar] [CrossRef]
- Mason, R.P.; Walter Mary, F.; Jacob Robert, F. Effects of HMG-CoA Reductase Inhibitors on Endothelial Function. Circulation 2004, 109, II-34–II-41. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.K. Molecular Clues into the Pathogenesis of Statin-Mediated Muscle Toxicity. Muscle Nerve 2005, 31, 572–580. [Google Scholar] [CrossRef]
- Nakahara, K.; Yada, T.; Kuriyama, M.; Osame, M. Cytosolic Ca2+ Increase and Cell Damage in L6 Rat Myoblasts by HMG-CoA Reductase Inhibitors. Biochem. Biophys. Res. Commun. 1994, 202, 1579–1585. [Google Scholar] [CrossRef]
- Kato, K.; Cox, A.D.; Hisaka, M.M.; Graham, S.M.; Buss, J.E.; Der, C.J. Isoprenoid Addition to Ras Protein Is the Critical Modification for Its Membrane Association and Transforming Activity. Proc. Natl. Acad. Sci. USA 1992, 89, 6403–6407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.K. Isoprenoids as Mediators of the Biological Effects of Statins. J. Clin. Investig. 2002, 110, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Trafficking and Signaling by Fatty-Acylated and Prenylated Proteins. Nat. Chem. Biol. 2006, 2, 584–590. [Google Scholar] [CrossRef]
- Epstein, W.W.; Lever, D.; Leining, L.M.; Bruenger, E.; Rilling, H.C. Quantitation of Prenylcysteines by a Selective Cleavage Reaction. Proc. Natl. Acad. Sci. USA 1991, 88, 9668–9670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikitake, Y.; Liao James, K. Rho GTPases, Statins, and Nitric Oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Paganelli, F.; Giuntini, G.; Mattioli, E.; Cappellini, A.; Ramazzotti, G.; Faenza, I.; Maltarello, M.C.; Martelli, A.M.; Scotlandi, K.; et al. Lamin A and Prelamin A Counteract Migration of Osteosarcoma Cells. Cells 2020, 9, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Casey, P.J. Protein Prenylation: Unique Fats Make Their Mark on Biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Sebti, S.M. Protein Farnesylation: Implications for Normal Physiology, Malignant Transformation, and Cancer Therapy. Cancer Cell 2005, 7, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Mullen, P.J.; Lüscher, B.; Scharnagl, H.; Krähenbühl, S.; Brecht, K. Effect of Simvastatin on Cholesterol Metabolism in C2C12 Myotubes and HepG2 Cells, and Consequences for Statin-Induced Myopathy. Biochem. Pharmacol. 2010, 79, 1200–1209. [Google Scholar] [CrossRef]
- Young, A.; Lou, D.; McCormick, F. Oncogenic and Wild-Type Ras Play Divergent Roles in the Regulation of Mitogen-Activated Protein Kinase Signaling. Cancer Discov. 2013, 3, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Zhong, Z.; Wang, F.; Xu, J.; Xu, F.; Kong, W.; Ling, Z.; Shu, N.; Li, Y.; Wu, T.; et al. Atorvastatin Impaired Glucose Metabolism in C2C12 Cells Partly via Inhibiting Cholesterol-Dependent Glucose Transporter 4 Translocation. Biochem. Pharmacol. 2018, 150, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Siddals, K.W.; Marshman, E.; Westwood, M.; Gibson, J.M. Abrogation of Insulin-like Growth Factor-I (IGF-I) and Insulin Action by Mevalonic Acid Depletion: Synergy between Protein Prenylation and Receptor Glycosylation Pathways. J. Biol. Chem. 2004, 279, 38353–38359. [Google Scholar] [CrossRef] [Green Version]
- Vaklavas, C.; Chatzizisis, Y.S.; Ziakas, A.; Zamboulis, C.; Giannoglou, G.D. Molecular Basis of Statin-Associated Myopathy. Atherosclerosis 2009, 202, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Marcoff, L.; Thompson, P.D. The Role of Coenzyme Q10 in Statin-Associated Myopathy: A Systematic Review. J. Am. Coll. Cardiol. 2007, 49, 2231–2237. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The Role of Mitochondria in Statin-Induced Myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernster, L.; Dallner, G. Biochemical, Physiological and Medical Aspects of Ubiquinone Function. Biochim. Et. Biophys. Acta Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Laaksonen, R.; Ojala, J.-P.; Tikkanen, M.J.; Himberg, J.-J. Serum Ubiquinone Concentrations after Short- and Long-Term Treatment with HMG-CoA Reductase Inhibitors. Eur. J. Clin. Pharm. 1994, 46, 313–317. [Google Scholar] [CrossRef]
- De Pinieux, G.; Chariot, P.; Ammi-Saïd, M.; Louarn, F.; Lejonc, J.L.; Astier, A.; Jacotot, B.; Gherardi, R. Lipid-Lowering Drugs and Mitochondrial Function: Effects of HMG-CoA Reductase Inhibitors on Serum Ubiquinone and Blood Lactate/Pyruvate Ratio. Br. J. Clin. Pharmacol. 1996, 42, 333–337. [Google Scholar] [CrossRef] [Green Version]
- Lotteau, S.; Ivarsson, N.; Yang, Z.; Restagno, D.; Colyer, J.; Hopkins, P.; Weightman, A.; Himori, K.; Yamada, T.; Bruton, J.; et al. A Mechanism for Statin-Induced Susceptibility to Myopathy. JACC Basic Transl. Sci. 2019, 4, 509–523. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium Signalling and Cell Proliferation. BioEssays 1995, 17, 491–500. [Google Scholar] [CrossRef]
- McConkey, D.J.; Orrenius, S. The Role of Calcium in the Regulation of Apoptosis. Biochem. Biophys. Res. Commun. 1997, 239, 357–366. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal Calcium Signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Mariot, P.; Prevarskaya, N.; Roudbaraki, M.M.; Bourhis, X.L.; Coppenolle, F.V.; Vanoverberghe, K.; Skryma, R. Evidence of Functional Ryanodine Receptor Involved in Apoptosis of Prostate Cancer (LNCaP) Cells. Prostate 2000, 43, 205–214. [Google Scholar] [CrossRef]
- Venturi, E.; Lindsay, C.; Lotteau, S.; Yang, Z.; Steer, E.; Witschas, K.; Wilson, A.D.; Wickens, J.R.; Russell, A.J.; Steele, D.; et al. Simvastatin Activates Single Skeletal RyR1 Channels but Exerts More Complex Regulation of the Cardiac RyR2 Isoform: Simvastatin Modulates RyR1 and RyR2 Channel Gating. Br. J. Pharmacol. 2018, 175, 938–952. [Google Scholar] [CrossRef] [Green Version]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity: Mechanistic Insights and Clinical Implications. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Ruaño, G.; Thompson, P.D.; Windemuth, A.; Seip, R.L.; Dande, A.; Sorokin, A.; Kocherla, M.; Smith, A.; Holford, T.R.; Wu, A.H.B. Physiogenomic Association of Statin-Related Myalgia to Serotonin Receptors. Muscle Nerve 2007, 36, 329–335. [Google Scholar] [CrossRef]
- Marciante, K.D.; Durda, J.P.; Heckbert, S.R.; Lumley, T.; Rice, K.; McKnight, B.; Totah, R.A.; Tamraz, B.; Kroetz, D.L.; Fukushima, H.; et al. Cerivastatin, Genetic Variants, and the Risk of Rhabdomyolysis. Pharm. Genom. 2011, 21, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Bitzur, R.; Cohen, H.; Kamari, Y.; Harats, D. Intolerance to Statins: Mechanisms and Management. Diabetes Care 2013, 36 (Suppl. S2), S325–S330. [Google Scholar] [CrossRef] [Green Version]
- März, W.; Laufs, U. Leucocyte Immunoglobulin-like Receptor Subfamily-B5 (LILRB5) Genetic Variation and Statin-Associated Muscle Symptoms: Another Piece in a Puzzling Puzzle. Eur. Heart J. 2017, 38, 3576–3578. [Google Scholar] [CrossRef]
- Siddiqui, M.K.; Maroteau, C.; Veluchamy, A.; Tornio, A.; Tavendale, R.; Carr, F.; Abelega, N.-U.; Carr, D.; Bloch, K.; Hallberg, P.; et al. A Common Missense Variant of LILRB5 Is Associated with Statin Intolerance and Myalgia. Eur. Heart J. 2017, 38, 3569–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopewell, J.C.; Offer, A.; Haynes, R.; Bowman, L.; Li, J.; Chen, F.; Bulbulia, R.; Lathrop, M.; Baigent, C.; Landray, M.J.; et al. Independent Risk Factors for Simvastatin-Related Myopathy and Relevance to Different Types of Muscle Symptom. Eur. Heart J. 2020, 41, 3336–3342. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C.; Krieger, J.N.; McNaughton-Collins, M. SLCO1B1 Variants and Statin-Induced Myopathy—A Genomewide Study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Niemi, M. Transporter Pharmacogenetics and Statin Toxicity. Clin. Pharm. 2010, 87, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Gandra, S.R.; McKendrick, J.; Dent, R.; Wieffer, H.; Cheng, L.-I.; Catapano, A.L.; Oh, P.; Kees Hovingh, G.; Stroes, E.S. Identification and Management of Statin-Associated Symptoms in Clinical Practice: Extension of a Clinician Survey to 12 Further Countries. Cardiovasc. Drugs 2017, 31, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Tiniakou, E. Statin-Associated Autoimmune Myopathy: Current Perspectives. Clin. Risk Manag. 2020, 16, 483–492. [Google Scholar] [CrossRef]
- Benveniste, O.; Drouot, L.; Jouen, F.; Charuel, J.-L.; Bloch-Queyrat, C.; Behin, A.; Amoura, Z.; Marie, I.; Guiguet, M.; Eymard, B.; et al. Correlation of Anti–Signal Recognition Particle Autoantibody Levels with Creatine Kinase Activity in Patients with Necrotizing Myopathy. Arthritis Rheum. 2011, 63, 1961–1971. [Google Scholar] [CrossRef]
- Allenbach, Y.; Mammen, A.L.; Benveniste, O.; Stenzel, W.; Allenbach, Y.; Amato, A.; Aussey, A.; Benveniste, O.; De Bleecker, J.; de Groot, I.; et al. 224th ENMC International Workshop. Neuromuscul. Disord. 2018, 28, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Allenbach, Y.; Drouot, L.; Rigolet, A.; Charuel, J.L.; Jouen, F.; Romero, N.B.; Maisonobe, T.; Dubourg, O.; Behin, A.; Laforet, P.; et al. Anti-HMGCR Autoantibodies in European Patients With Autoimmune Necrotizing Myopathies: Inconstant Exposure to Statin. Medicine 2014, 93, 150–157. [Google Scholar] [CrossRef]
- Watanabe, Y.; Suzuki, S.; Nishimura, H.; Murata, K.; Kurashige, T.; Ikawa, M.; Asahi, M.; Konishi, H.; Mitsuma, S.; Kawabata, S.; et al. Statins and Myotoxic Effects Associated With Anti-3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Autoantibodies: An Observational Study in Japan. Medicine 2015, 94, e416. [Google Scholar] [CrossRef]
- Kadoya, M.; Hida, A.; Hashimoto Maeda, M.; Taira, K.; Ikenaga, C.; Uchio, N.; Kubota, A.; Kaida, K.; Miwa, Y.; Kurasawa, K.; et al. Cancer Association as a Risk Factor for Anti-HMGCR Antibody-Positive Myopathy. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Lu, X.; Peng, Q.; Shu, X.; Wang, G. Clinical Characteristics of Anti-3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Antibodies in Chinese Patients with Idiopathic Inflammatory Myopathies. PLoS ONE 2015, 10, e0141616. [Google Scholar] [CrossRef]
- Arouche-Delaperche, L.; Allenbach, Y.; Amelin, D.; Preusse, C.; Mouly, V.; Mauhin, W.; Tchoupou, G.D.; Drouot, L.; Boyer, O.; Stenzel, W.; et al. Pathogenic Role of Anti–Signal Recognition Protein and Anti–3-Hydroxy-3-Methylglutaryl-CoA Reductase Antibodies in Necrotizing Myopathies: Myofiber Atrophy and Impairment of Muscle Regeneration in Necrotizing Autoimmune Myopathies. Ann. Neurol. 2017, 81, 538–548. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of Action of Fibrates on Lipid and Lipoprotein Metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Hodel, C. Myopathy and Rhabdomyolysis with Lipid-Lowering Drugs. Toxicol. Lett. 2002, 128, 159–168. [Google Scholar] [CrossRef]
- Pruimboom-Brees, I.; Haghpassand, M.; Royer, L.; Brees, D.; Aldinger, C.; Reagan, W.; Singh, J.; Kerlin, R.; Kane, C.; Bagley, S.; et al. A Critical Role for Peroxisomal Proliferator-Activated Receptor-Alpha Nuclear Receptors in the Development of Cardiomyocyte Degeneration and Necrosis. Am. J. Pathol. 2006, 169, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilde, A.J.; van der Lee, K.A.J.M.; Willemsen, P.H.M.; Chinetti, G.; van der Leij, F.R.; van der Vusse, G.J.; Staels, B.; van Bilsen, M. Peroxisome Proliferator-Activated Receptor (PPAR) Alpha and PPARbeta/Delta, but Not PPARgamma, Modulate the Expression of Genes Involved in Cardiac Lipid Metabolism. Circ. Res. 2003, 92, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial Impairment by PPAR Agonists and Statins Identified via Immunocaptured OXPHOS Complex Activities and Respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, J.C.; Pruimboom-Brees, I.; Francone, O.L.; Amacher, D.E.; Boldt, S.E.; Kerlin, R.L.; Ballinger, W.E. The PPARα Agonists Fenofibrate and CP-778875 Cause Increased β-Oxidation, Leading to Oxidative Injury in Skeletal and Cardiac Muscle in the Rat. Toxicol. Pathol. 2012, 40, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motojima, K. Differential Effects of PPARα Activators on Induction of Ectopic Expression of Tissue-Specific Fatty Acid Binding Protein Genes in the Mouse Liver. Int. J. Biochem. Cell Biol. 2000, 32, 1085–1092. [Google Scholar] [CrossRef]
- Owczarek, J.; Jasińska, M.; Orszulak-Michalak, D. Drug-Induced Myopathies. An Overview of the Possible Mechanisms. Pharm. Rep. 2005, 57, 23–34. [Google Scholar]
- Shek, A.; Ferrill, M.J. Statin—Fibrate Combination Therapy. Ann. Pharm. 2001, 35, 908–917. [Google Scholar] [CrossRef]
- Wang, J.-S.; Wen, X.; Backman, J.T.; Neuvonen, P.J. Effect of Albumin and Cytosol on Enzyme Kinetics of Tolbutamide Hydroxylation and on Inhibition of CYP2C9 by Gemfibrozil in Human Liver Microsomes. J. Pharm. Exp. 2002, 302, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Prueksaritanont, T.; Tang, C.; Qiu, Y.; Mu, L.; Subramanian, R.; Lin, J.H. Effects of Fibrates on Metabolism of Statins in Human Hepatocytes. Drug Metab. Dispos. 2002, 30, 1280–1287. [Google Scholar] [CrossRef] [Green Version]
- Tojcic, J.; Benoit-Biancamano, M.-O.; Court, M.H.; Straka, R.J.; Caron, P.; Guillemette, C. In Vitro Glucuronidation of Fenofibric Acid by Human UDP-Glucuronosyltransferases and Liver Microsomes. Drug Metab. Dispos. 2009, 37, 2236–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.B.; Spence, J.D. Clinical Pharmacokinetics of Fibric Acid Derivatives (Fibrates). Clin. Pharm. 1998, 34, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Li, B.; Louie, S.W.; Pudvah, N.T.; Stocco, R.; Wong, W.; Abramovitz, M.; Demartis, A.; Laufer, R.; Hochman, J.H.; et al. Effects of Fibrates on Human Organic Anion-Transporting Polypeptide 1B1-, Multidrug Resistance Protein 2- and P-Glycoprotein-Mediated Transport. Xenobiotica 2005, 35, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Kawata, R.; Yokoi, T. Analysis of a Skeletal Muscle Injury and Drug Interactions in Lovastatin- and Fenofibrate-Coadministered Dogs. Int. J. Toxicol. 2019, 38, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Davis, H.R.; Zhu, L.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.N.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 Protein Is Critical for Intestinal Cholesterol Absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [Green Version]
- Davis, H.R.; Zhu, L.; Hoos, L.M.; Tetzloff, G.; Maguire, M.; Liu, J.; Yao, X.; Iyer, S.P.N.; Lam, M.-H.; Lund, E.G.; et al. Niemann-Pick C1 Like 1 (NPC1L1) Is the Intestinal Phytosterol and Cholesterol Transporter and a Key Modulator of Whole-Body Cholesterol Homeostasis. J. Biol. Chem. 2004, 279, 33586–33592. [Google Scholar] [CrossRef] [Green Version]
- Florentin, M.; Liberopoulos, E.N.; Elisaf, M.S. Ezetimibe-Associated Adverse Effects: What the Clinician Needs to Know: Ezetimibe and Side Effects. Int. J. Clin. Pract. 2007, 62, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Perez-Calvo, J.; Civeira-Murillo, F.; Cabello, A. Worsening Myopathy Associated with Ezetimibe in a Patient with McArdle Disease. QJM Int. J. Med. 2005, 98, 461–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havranek, J.M.; Wolfsen, A.R.; Warnke, G.A.; Phillips, P.S. Monotherapy with Ezetimibe Causing Myopathy. Am. J. Med. 2006, 119, 285–286. [Google Scholar] [CrossRef]
- Simard, C.; Poirier, P. Ezetimibe-Associated Myopathy in Monotherapy and in Combination with a 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitor. Can. J. Cardiol. 2006, 22, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Weffald, L.A.; Flach, L.A. Myopathy Associated with Atorvastatin-Ezetimibe Combination Therapy. Pharmacotherapy 2007, 27, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Fux, R.; Mörike, K.; Gundel, U.-F.; Hartmann, R.; Gleiter, C.H. Ezetimibe and Statin-Associated Myopathy. Ann. Intern. Med. 2004, 140, 671–672. [Google Scholar] [CrossRef]
- Simard, C.; Turgeon, J. The Pharmacokinetics of Ezetimibe. Can. J. Clin. Pharm. 2003, 10 (Suppl. A), 13A–20A. [Google Scholar]
- Hsiang, B.; Zhu, Y.; Wang, Z.; Wu, Y.; Sasseville, V.; Yang, W.-P.; Kirchgessner, T.G. A Novel Human Hepatic Organic Anion Transporting Polypeptide (OATP2). J. Biol. Chem. 1999, 274, 37161–37168. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, V.; Moja, L.; Dall’Olmo, L.; Cappellini, G.; Garattini, S. Most Appropriate Animal Models to Study the Efficacy of Statins: A Systematic Review. Eur. J. Clin. Investig. 2014, 44, 848–871. [Google Scholar] [CrossRef]
- Jaśkiewicz, A.; Pająk, B.; Łabieniec-Watała, M.; Palma, C.D.; Orzechowski, A. Diverse Action of Selected Statins on Skeletal Muscle Cells—An Attempt to Explain the Protective Effect of Geranylgeraniol (GGOH) in Statin-Associated Myopathy (SAM). J. Clin. Med. 2019, 8, 694. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.C.; Fenning, A.S.; Vella, R.K. Geranylgeraniol Prevents Statin-Induced Skeletal Muscle Fatigue without Causing Adverse Effects in Cardiac or Vascular Smooth Muscle Performance. Transl. Res. 2020, 215, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, S.A.; Popp, O.; Haafke, S.; Jedraszczak, N.; Grieben, U.; Saar, K.; Patone, G.; Kress, W.; Steinhagen-Thiessen, E.; Dittmar, G.; et al. Statin-Induced Myopathic Changes in Primary Human Muscle Cells and Reversal by a Prostaglandin F2 Alpha Analogue. Sci. Rep. 2020, 10, 2158. [Google Scholar] [CrossRef] [Green Version]
- Madden, L.; Juhas, M.; Kraus, W.E.; Truskey, G.A.; Bursac, N. Bioengineered Human Myobundles Mimic Clinical Responses of Skeletal Muscle to Drugs. eLife 2015, 4, e04885. [Google Scholar] [CrossRef] [Green Version]
- Ananthakumar, A.; Liu, Y.; Fernandez, C.E.; Truskey, G.A.; Voora, D. Modeling Statin Myopathy in a Human Skeletal Muscle Microphysiological System. PLoS ONE 2020, 15, e0242422. [Google Scholar] [CrossRef]
- Osaki, Y.; Nakagawa, Y.; Miyahara, S.; Iwasaki, H.; Ishii, A.; Matsuzaka, T.; Kobayashi, K.; Yatoh, S.; Takahashi, A.; Yahagi, N.; et al. Skeletal Muscle-Specific HMG-CoA Reductase Knockout Mice Exhibit Rhabdomyolysis: A Model for Statin-Induced Myopathy. Biochem. Biophys. Res. Commun. 2015, 466, 536–540. [Google Scholar] [CrossRef]
- Grajales-Reyes, G.E.; Báez-Pagán, C.A.; Zhu, H.; Grajales-Reyes, J.G.; Delgado-Vélez, M.; García-Beltrán, W.F.; Luciano, C.A.; Quesada, O.; Ramírez, R.; Gómez, C.M.; et al. Transgenic Mouse Model Reveals an Unsuspected Role of the Acetylcholine Receptor in Statin-Induced Neuromuscular Adverse Drug Reactions. Pharm. J. 2013, 13, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Reijneveld, J.C.; Koot, R.W.; Bredman, J.J.; Joles, J.A.; Bär, P.R. Differential Effects of 3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Inhibitors on the Development of Myopathy in Young Rats. Pediatr. Res. 1996, 39, 1028–1035. [Google Scholar] [CrossRef] [Green Version]
- Westwood, F.R.; Bigley, A.; Randall, K.; Marsden, A.M.; Scott, R.C. Statin-Induced Muscle Necrosis in the Rat: Distribution, Development, and Fibre Selectivity. Toxicol. Pathol. 2005, 33, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obayashi, H.; Nezu, Y.; Yokota, H.; Kiyosawa, N.; Mori, K.; Maeda, N.; Tani, Y.; Manabe, S.; Sanbuissho, A. Cerivastatin Induces Type-I Fiber-, Not Type-II Fiber-, Predominant Muscular Toxicity in the Young Male F344 Rats. J. Toxicol. Sci. 2011, 36, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallinson, J.E.; Constantin-Teodosiu, D.; Sidaway, J.; Westwood, F.R.; Greenhaff, P.L. Blunted Akt/FOXO Signalling and Activation of Genes Controlling Atrophy and Fuel Use in Statin Myopathy: Blunted Akt/FOXO Signalling in Statin-Induced Myopathy. J. Physiol. 2009, 587, 219–230. [Google Scholar] [CrossRef]
- Camerino, G.M.; Pellegrino, M.A.; Brocca, L.; Digennaro, C.; Camerino, D.C.; Pierno, S.; Bottinelli, R. Statin or Fibrate Chronic Treatment Modifies the Proteomic Profile of Rat Skeletal Muscle. Biochem. Pharm. 2011, 81, 1054–1064. [Google Scholar] [CrossRef] [Green Version]
- Fukami, M.; Maeda, N.; Fukushige, J.; Kogure, Y.; Shimada, Y.; Ogawa, T.; Tsujita, Y. Effects of HMG-CoA Reductase Inhibitors on Skeletal Muscles of Rabbits. Res. Exp. Med. 1993, 193, 263–273. [Google Scholar] [CrossRef]
- Nakahara, K.; Kuriyama, M.; Sonoda, Y.; Yoshidome, H.; Nakagawa, H.; Fujiyama, J.; Higuchi, I.; Osame, M. Myopathy Induced by HMG-CoA Reductase Inhibitors in Rabbits: A Pathological, Electrophysiological, and Biochemical Study. Toxicol. Appl. Pharm. 1998, 152, 99–106. [Google Scholar] [CrossRef]
- Mohd Azlan, P.; Jahromi, M.F.; Ariff, M.O.; Ebrahimi, M.; Candyrine, S.C.L.; Liang, J.B. Aspergillus Terreus Treated Rice Straw Suppresses Methane Production and Enhances Feed Digestibility in Goats. Trop. Anim. Health Prod. 2018, 50, 565–571. [Google Scholar] [CrossRef]
- Leo, T.K.; Garba, S.; Abubakar, D.; Sazili, A.Q.; Candyrine, S.C.L.; Jahromi, M.F.; Goh, Y.M.; Ronimus, R.; Muetzel, S.; Liang, J.B. Naturally Produced Lovastatin Modifies the Histology and Proteome Profile of Goat Skeletal Muscle. Animals 2019, 10, 72. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, N.P.; Kim, M.J.; Bible, K.L.; Adams, M.E.; Froehner, S.C. A New Therapeutic Effect of Simvastatin Revealed by Functional Improvement in Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 12864–12869. [Google Scholar] [CrossRef] [Green Version]
- Bandaru, M.K.; Emmanouilidou, A.; Ranefall, P.; von der Heyde, B.; Mazzaferro, E.; Klingström, T.; Masiero, M.; Dethlefsen, O.; Ledin, J.; Larsson, A.; et al. Zebrafish Larvae as a Model. System for Systematic Characterization of Drugs and Genes in Dyslipidemia and Atherosclerosis. Genomics 2018, preprint. [Google Scholar] [CrossRef]
- Liu, C.; Kim, Y.S.; Kim, J.; Pattison, J.; Kamaid, A.; Miller, Y.I. Modeling Hypercholesterolemia and Vascular Lipid Accumulation in LDL Receptor Mutant Zebrafish. J. Lipid Res. 2018, 59, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, E.M.; Horstick, E.J.; Dowling, J.J. Swimming into Prominence: The Zebrafish as a Valuable Tool for Studying Human Myopathies and Muscular Dystrophies. FEBS J. 2013, 280, 4187–4197. [Google Scholar] [CrossRef] [Green Version]
- Plantié, E.; Migocka-Patrzałek, M.; Daczewska, M.; Jagla, K. Model Organisms in the Fight against Muscular Dystrophy: Lessons from Drosophila and Zebrafish. Molecules 2015, 20, 6237–6253. [Google Scholar] [CrossRef] [Green Version]
- Dubińska-Magiera, M.; Daczewska, M.; Lewicka, A.; Migocka-Patrzałek, M.; Niedbalska-Tarnowska, J.; Jagla, K. Zebrafish: A Model for the Study of Toxicants Affecting Muscle Development and Function. Int. J. Mol. Sci. 2016, 17, 1941. [Google Scholar] [CrossRef]
- Thorpe, J.L.; Doitsidou, M.; Ho, S.-Y.; Raz, E.; Farber, S.A. Germ Cell Migration in Zebrafish Is Dependent on HMGCoA Reductase Activity and Prenylation. Dev. Cell 2004, 6, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Hanai, J.; Cao, P.; Tanksale, P.; Imamura, S.; Koshimizu, E.; Zhao, J.; Kishi, S.; Yamashita, M.; Phillips, P.S.; Sukhatme, V.P.; et al. The Muscle-Specific Ubiquitin Ligase Atrogin-1/MAFbx Mediates Statin-Induced Muscle Toxicity. J. Clin. Investig. 2007, JCI32741. [Google Scholar] [CrossRef]
- Campos, L.M.; Rios, E.A.; Midlej, V.; Atella, G.C.; Herculano-Houzel, S.; Benchimol, M.; Mermelstein, C.; Costa, M.L. Structural Analysis of Alterations in Zebrafish Muscle Differentiation Induced by Simvastatin and Their Recovery with Cholesterol. J. Histochem. Cytochem. 2015, 63, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, S.; Torres, T.; Martins, R.; Santos, M.M. Toxicity Screening of Diclofenac, Propranolol, Sertraline and Simvastatin Using Danio Rerio and Paracentrotus Lividus Embryo Bioassays. Ecotoxicol. Environ. Saf. 2015, 114, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Valbuena Perez, J.V.; Linnenberger, R.; Dahlem, C.; Legroux, T.M.; Hecksteden, A.; Tse, W.K.F.; Flamini, S.; Andreas, A.; Herrmann, J.; et al. The Glucocorticoid-induced Leucine Zipper Mediates Statin-induced Muscle Damage. FASEB J. 2020, 34, 4684–4701. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, K.; Giesy, J.P.; Hu, J. Families of Nuclear Receptors in Vertebrate Models: Characteristic and Comparative Toxicological Perspective. Sci. Rep. 2015, 5, 8554. [Google Scholar] [CrossRef] [Green Version]
- Barros, S.; Montes, R.; Quintana, J.B.; Rodil, R.; André, A.; Capitão, A.; Soares, J.; Santos, M.M.; Neuparth, T. Chronic Environmentally Relevant Levels of Simvastatin Induces Non-Monotonic Responses in Zebrafish (Danio Rerio). Pharmacol. Toxicol. 2018. preprint. [Google Scholar] [CrossRef] [Green Version]
- Coimbra, A.M.; Peixoto, M.J.; Coelho, I.; Lacerda, R.; Carvalho, A.P.; Gesto, M.; Lyssimachou, A.; Lima, D.; Soares, J.; André, A.; et al. Chronic Effects of Clofibric Acid in Zebrafish (Danio Rerio): A Multigenerational Study. Aquat. Toxicol. 2015, 160, 76–86. [Google Scholar] [CrossRef]
- Raldua, D.; Campos, B.; Barata, C.; Piña, B.; García-Reyero, N.; Babin, P.J. Deciphering Emerging Toxicological Effects of Pharmaceuticals on Aquatic Organisms by Using Daphnia Magna and Danio Rerio as Model Organisms. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2013; Volume 62, pp. 611–647. [Google Scholar] [CrossRef]
- Baek, J.S.; Fang, L.; Li, A.C.; Miller, Y.I. Ezetimibe and Simvastatin Reduce Cholesterol Levels in Zebrafish Larvae Fed a High-Cholesterol Diet. Cholesterol 2012, 2012, 564705. [Google Scholar] [CrossRef] [Green Version]
- Bruscoli, S.; Donato, V.; Velardi, E.; Di Sante, M.; Migliorati, G.; Donato, R.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper (GILZ) and Long GILZ Inhibit Myogenic Differentiation and Mediate Anti-Myogenic Effects of Glucocorticoids. J. Biol. Chem. 2010, 285, 10385–10396. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Liu, J.-X.; Hu, B.; Xiao, W. Zebrafish Foxo3b Negatively Regulates Canonical Wnt Signaling to Affect Early Embryogenesis. PLoS ONE 2011, 6, e24469. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a Muscle-Specific F-Box Protein Highly Expressed during Muscle Atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, P.; Hanai, J.; Tanksale, P.; Imamura, S.; Sukhatme, V.P.; Lecker, S.H. Statin-induced Muscle Damage and Atrogin-1 Induction Is the Result of a Geranylgeranylation Defect. FASEB J. 2009, 23, 2844–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, B.K.; Gilbert, T.J.; Hanai, J.; Imamura, S.; Bodycombe, N.E.; Bon, R.S.; Waldmann, H.; Clemons, P.A.; Sukhatme, V.P.; Mootha, V.K. A Small-Molecule Screening Strategy to Identify Suppressors of Statin Myopathy. ACS Chem. Biol. 2011, 6, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Campos, L.M.; Rios, E.A.; Guapyassu, L.; Midlej, V.; Atella, G.C.; Herculano-Houzel, S.; Benchimol, M.; Mermelstein, C.; Costa, M.L. Alterations in Zebrafish Development Induced by Simvastatin: Comprehensive Morphological and Physiological Study, Focusing on Muscle. Exp. Biol. Med. 2016, 241, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Lamperti, C.; Naini, A.B.; Lucchini, V.; Prelle, A.; Bresolin, N.; Moggio, M.; Sciacco, M.; Kaufmann, P.; DiMauro, S. Muscle Coenzyme Q10 Level in Statin-Related Myopathy. Arch. Neurol. 2005, 62, 1709–1712. [Google Scholar] [CrossRef] [Green Version]
- Pasha, R.; Moon, T.W. Coenzyme Q10 Protects against Statin-Induced Myotoxicity in Zebrafish Larvae ( Danio Rerio ). Environ. Toxicol. Pharmacol. 2017, 52, 150–160. [Google Scholar] [CrossRef]
- Chen, J.W.; Niu, X.; King, M.J.; Noedl, M.-T.; Tabin, C.J.; Galloway, J.L. The Mevalonate Pathway Is a Crucial Regulator of Tendon Cell Specification. Development 2020, 147, dev185389. [Google Scholar] [CrossRef]
- Backes, J.M.; Gibson, C.A.; Ruisinger, J.F.; Moriarty, P.M. Fibrates: What Have We Learned in the Past 40 Years? Pharmacotherapy 2007, 27, 412–424. [Google Scholar] [CrossRef]
- Raldúa, D.; André, M.; Babin, P.J. Clofibrate and Gemfibrozil Induce an Embryonic Malabsorption Syndrome in Zebrafish. Toxicol. Appl. Pharmacol. 2008, 228, 301–314. [Google Scholar] [CrossRef]
- Ottmar, K.J.; Colosi, L.M.; Smith, J.A. Fate and Transport of Atorvastatin and Simvastatin Drugs during Conventional Wastewater Treatment. Chemosphere 2012, 88, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.M.; Ruivo, R.; Lopes-Marques, M.; Torres, T.; de los Santos, C.B.; Castro, L.F.C.; Neuparth, T. Statins: An Undesirable Class of Aquatic Contaminants? Aquat. Toxicol. 2016, 174, 1–9. [Google Scholar] [CrossRef]
- Togola, A.; Budzinski, H. Analytical Development for Analysis of Pharmaceuticals in Water Samples by SPE and GC–MS. Anal. Bioanal. Chem. 2007, 388, 627–635. [Google Scholar] [CrossRef]
- Wu, Q.; Shi, H.; Adams, C.D.; Timmons, T.; Ma, Y. Oxidative Removal of Selected Endocrine-Disruptors and Pharmaceuticals in Drinking Water Treatment Systems, and Identification of Degradation Products of Triclosan. Sci. Total Environ. 2012, 439, 18–25. [Google Scholar] [CrossRef]
- Hernando, M.D.; Agüera, A.; Fernández-Alba, A.R. LC-MS Analysis and Environmental Risk of Lipid Regulators. Anal. Bioanal. Chem. 2007, 387, 1269–1285. [Google Scholar] [CrossRef]
- Arnold, K.E.; Brown, A.R.; Ankley, G.T.; Sumpter, J.P. Medicating the Environment: Assessing Risks of Pharmaceuticals to Wildlife and Ecosystems. Philos. Trans. R. Soc. B 2014, 369, 20130569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingcade, A.; Ahuja, N.; Jefferson, A.; Schaffer, P.A.; Ryschon, H.; Cadmus, P.; Garrity, D.; Ramsdell, H. Morbidity and Mortality in Danio Rerio and Pimephales Promelas Exposed to Antilipidemic Drug Mixtures (Fibrates and Statins) during Embryogenesis: Comprehensive Assessment via Ante and Post Mortem Endpoints. Chemosphere 2021, 263, 127911. [Google Scholar] [CrossRef]
- Cunha, V.; Santos, M.M.; Moradas-Ferreira, P.; Ferreira, M. Simvastatin Effects on Detoxification Mechanisms in Danio Rerio Embryos. Environ. Sci. Pollut. Res. Int. 2016, 23, 10615–10629. [Google Scholar] [CrossRef]
- Cunha, V.; Santos, M.M.; Moradas-Ferreira, P.; Castro, L.F.C.; Ferreira, M. Simvastatin Modulates Gene Expression of Key Receptors in Zebrafish Embryos. J. Toxicol. Environ. Health Part A 2017, 80, 465–476. [Google Scholar] [CrossRef]
- Gronemeyer, H.; Gustafsson, J.-A.; Laudet, V. Principles for Modulation of the Nuclear Receptor Superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef] [PubMed]
- De Sotomayor, M.A.; Vega, S.; Mingorance, C.; Marhuenda, E.; Herrera, M.D. Effects of HMG-CoA Reductase Inhibition by Simvastatin on Vascular Dysfunction Induced by Lipopolysaccharide in Rats. Pharmacology 2008, 82, 89–96. [Google Scholar] [CrossRef]
- Fraunberger, P.; Gröne, E.; Gröne, H.-J.; Walli, A.K. Simvastatin Reduces Endotoxin-Induced Nuclear Factor KappaB Activation and Mortality in Guinea Pigs despite Lowering Circulating Low-Density Lipoprotein Cholesterol. Shock 2009, 32, 159–163. [Google Scholar] [CrossRef]
- Tete, V.S.; Nyoni, H.; Mamba, B.B.; Msagati, T.A.M. Occurrence and Spatial Distribution of Statins, Fibrates and Their Metabolites in Aquatic Environments. Arab. J. Chem. 2020, 13, 4358–4373. [Google Scholar] [CrossRef]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widrick, J.J.; Kawahara, G.; Alexander, M.S.; Beggs, A.H.; Kunkel, L.M. Discovery of Novel Therapeutics for Muscular Dystrophies Using Zebrafish Phenotypic Screens. J. Neuromuscul. Dis. 2019, 6, 271–287. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.M.; Hernandez, J.A.; Gonzalez, C.G. Functional Analysis of the Zebrafish Ortholog of HMGCS1 Reveals Independent Functions for Cholesterol and Isoprenoids in Craniofacial Development. PLoS ONE 2017, 12, e0180856. [Google Scholar] [CrossRef] [Green Version]
- ZFIN. Available online: http://zfin.org/ (accessed on 15 April 2021).
- The Alliance of Genome Resources Consortium; Agapite, J.; Albou, L.-P.; Aleksander, S.; Argasinska, J.; Arnaboldi, V.; Attrill, H.; Bello, S.M.; Blake, J.A.; Blodgett, O.; et al. Alliance of Genome Resources Portal: Unified Model Organism Research Platform. Nucleic Acids Res. 2020, 48, D650–D658. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl Variation Resources. Database 2018, 2018. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Human Protein Atlas. Available online: http://www.proteinatlas.org (accessed on 15 April 2021).
- D’Amico, L.; Scott, I.C.; Jungblut, B.; Stainier, D.Y.R. A Mutation in Zebrafish Hmgcr1b Reveals a Role for Isoprenoids in Vertebrate Heart-Tube Formation. Curr. Biol. 2007, 17, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Mapp, O.M.; Walsh, G.S.; Moens, C.B.; Tada, M.; Prince, V.E. Zebrafish Prickle1b Mediates Facial Branchiomotor Neuron Migration via a Farnesylation-Dependent Nuclear Activity. Development 2011, 138, 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Eisa-Beygi, S.; Hatch, G.; Noble, S.; Ekker, M.; Moon, T.W. The 3-Hydroxy-3-Methylglutaryl-CoA Reductase (HMGCR) Pathway Regulates Developmental Cerebral-Vascular Stability via Prenylation-Dependent Signalling Pathway. Dev. Biol. 2013, 373, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Barman, A.; Deb, B.; Chakraborty, S. A Glance at Genome Editing with CRISPR–Cas9 Technology. Curr. Genet. 2020, 66, 447–462. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR Web Toolbox beyond Genome Editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [Green Version]
- Kroll, F.; Powell, G.T.; Ghosh, M.; Gestri, G.; Antinucci, P.; Hearn, T.J.; Tunbak, H.; Lim, S.; Dennis, H.W.; Fernandez, J.M.; et al. A Simple and Effective F0 Knockout Method for Rapid Screening of Behaviour and Other Complex Phenotypes. eLife 2021, 10, e59683. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Karpf, J.A.; Myers, J.A.; Alexander, M.S.; Guyon, J.R.; Kunkel, L.M. Drug Screening in a Zebrafish Model of Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2011, 108, 5331–5336. [Google Scholar] [CrossRef] [Green Version]
- Waugh, T.A.; Horstick, E.; Hur, J.; Jackson, S.W.; Davidson, A.E.; Li, X.; Dowling, J.J. Fluoxetine Prevents Dystrophic Changes in a Zebrafish Model of Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2014, 23, 4651–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, P.R.; Feyder, M.J.; Hightower, R.M.; Garcia-Perez, D.; Vieira, N.M.; Lek, A.; Gibbs, D.E.; Moukha-Chafiq, O.; Augelli-Szafran, C.E.; Kawahara, G.; et al. A Limb-Girdle Muscular Dystrophy 2I Model of Muscular Dystrophy Identifies Corrective Drug Compounds for Dystroglycanopathies. JCI Insight 2018, 3, e120493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Sztal, T.; Currie, P.D. Quantification of Birefringence Readily Measures the Level of Muscle Damage in Zebrafish. Biochem. Biophys. Res. Commun. 2012, 423, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Montandon, M.; Currie, P.D.; Ruparelia, A.A. Examining Muscle Regeneration in Zebrafish Models of Muscle Disease. JoVE 2021, 167, 62071. [Google Scholar] [CrossRef]
- Smith, S.J.; Horstick, E.J.; Davidson, A.E.; Dowling, J. Analysis of Zebrafish Larvae Skeletal Muscle Integrity with Evans Blue Dye. JoVE 2015, 105, 53183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.E.; Wood, A.J.; Ehrlich, O.; Li, M.; Sonntag, C.S.; Cole, N.J.; Huttner, I.G.; Sztal, T.E.; Currie, P.D. Cellular Rescue in a Zebrafish Model of Congenital Muscular Dystrophy Type 1A. NPJ Regen. Med. 2019, 4, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, S.E.; Miller, A.L. Visualization of Ca2+ Signaling During Embryonic Skeletal Muscle Formation in Vertebrates. Cold Spring Harb. Perspect. Biol. 2011, 3, a004325. [Google Scholar] [CrossRef]
- Sztal, T.E.; Ruparelia, A.A.; Williams, C.; Bryson-Richardson, R.J. Using Touch-Evoked Response and Locomotion Assays to Assess Muscle Performance and Function in Zebrafish. J. Vis. Exp. 2016, 116, 54431. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Li, G.; Xiao, Q.; Jiang, H.-R.; Tchivelekete, G.M.; Shu, X.; Liu, H. Quantification of the Influence of Drugs on Zebrafish Larvae Swimming Kinematics and Energetics. PeerJ 2020, 8, e8374. [Google Scholar] [CrossRef]
- Mead, A.F.; Kennedy, G.G.; Palmer, B.M.; Ebert, A.M.; Warshaw, D.M. Mechanical Characteristics of Ultrafast Zebrafish Larval Swimming Muscles. Biophys. J. 2020, 119, 806–820. [Google Scholar] [CrossRef]
- Voesenek, C.J.; Li, G.; Muijres, F.T.; van Leeuwen, J.L. Experimental–Numerical Method for Calculating Bending Moments in Swimming Fish Shows That Fish Larvae Control Undulatory Swimming with Simple Actuation. PLoS Biol. 2020, 18, e3000462. [Google Scholar] [CrossRef]
- Mitchell, C.; Caroff, L.; Solis-Lemus, J.A.; Reyes-Aldasoro, C.C.; Vigilante, A.; Warburton, F.; de Chaumont, F.; Dufour, A.; Dallongeville, S.; Olivo-Marin, J.-C.; et al. Cell Tracking Profiler—A User-Driven Analysis Framework for Evaluating 4D Live-Cell Imaging Data. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Chin, J.S.R.; Albert, L.T.; Loomis, C.L.; Keene, A.C.; Duboué, E.R. Behavioral Approaches to Studying Innate Stress in Zebrafish. J. Vis. Exp. 2019, 147, 59092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henn, K.; Braunbeck, T. Dechorionation as a Tool to Improve the Fish Embryo Toxicity Test (FET) with the Zebrafish (Danio Rerio). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2011, 153, 91–98. [Google Scholar] [CrossRef] [PubMed]
- De Abreu, M.S.; Kalueff, A.V. Of Mice and Zebrafish: The Impact of the Experimenter Identity on Animal Behavior. Lab. Anim. 2021, 50, 7. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal Models of Human Disease: Zebrafish Swim into View. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]


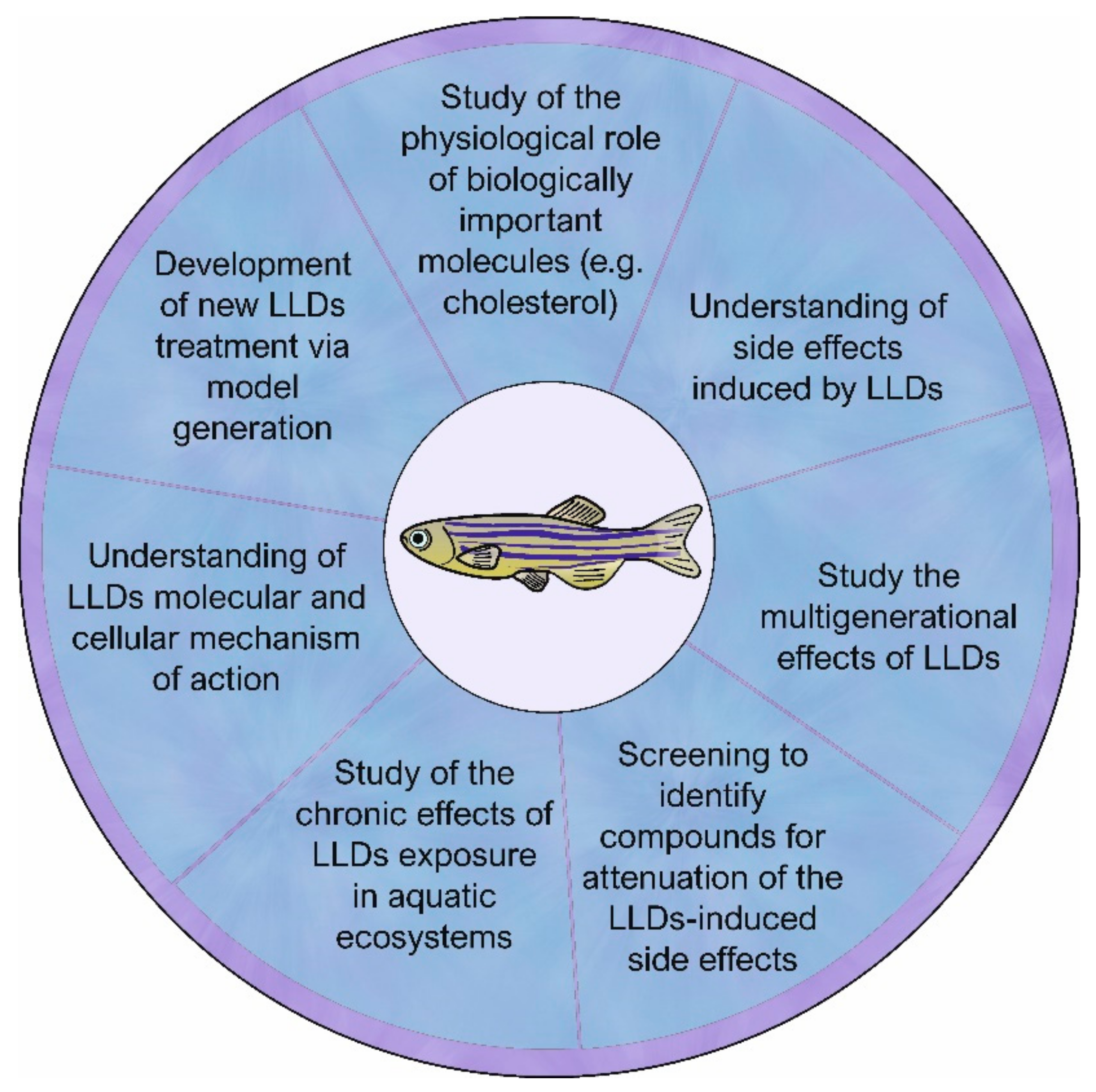{kind=link}
| Compound | Model | Outcome | Reference |
|---|---|---|---|
| Statins | |||
| Atorvastatin (ATV) | in vitro | Decrease of cholesterol level in C2C12 cells, impairment the translocation and function of glucose transporter GLUT4, less myotoxicity, impaired cellular mitochondrial respiration. | [8,32,91] |
| zebrafish | Embryos: germ cell migration defects and mild morphologic abnormalities. | [112] | |
| rat | Down-regulation of protein expression (proteins associated with energy production systems (including oxidative and glycolytic enzymes and CK), heat shock proteins, and proteins being components of myofibrils | [101] | |
| Fluvastatin (FLV) | rat | No significant alterations of proteins expression involved in energy production systems, overexpression of chaperonin 60, down-regulation of myozenin 1, FLV high FLV dose -increase the plasma CK content, lower FLV dose—no effects of CK content | [101] |
| Lovastatin (LOV) | zebrafish | Embryos: stimulation of atrogin-1expression, muscle fibre damage, developmental arrest, improper axis elongation, compressed somites | [112,113] |
| rat | Skeletal muscle damage | [97] | |
| goat | High CK activity, myopathy, fibre necrosis, skeletal muscle damage | [105] | |
| dog | Elevated level of CK, skeletal muscle fibres necrosis | [78] | |
| Pravastatin (PRA) | rat | No change of cholesterol level, small muscle damage, smaller body size | [97] |
| Rosuvastatin (RSV) | in vitro | Reduction in cholesterol biosynthesis, disruption of muscle cells differentiation and proliferation, changes in profiles of eicosanoids | [92] |
| Simvastin (SIM) | in vitro | Myotoxicity, impaired cellular mitochondrial respiration, reduction in cholesterol biosynthesis, disruption of muscle cells differentiation and proliferation, changes in profiles of eicosanoids | [91,92] |
| zebrafish | Adults: muscle structural damage, impaired movements and reduced heart beating, offspring embryonic malformations Embryos: changes in the muscle cytoskeleton, extracellular matrix, adhesion markers, and myofibrils organization, pericardial oedema, developmental arrest, improper axis elongation, compressed somites, transcription upregulation of ppars, pxr, and ahr, downregulation of pxr and ahr with no changes in ppars expression | [112,114,115,118] | |
| rat | Down-regulation PI3k/Akt signalling, and up-regulation FOXO transcription factors, an increase in the transcription of genes implicated in proteasomal- and lysosomal-mediated protein degradation (MAFbx), impairment of carbohydrate oxidation, oxidative stress, inflammation, an increased plasma CK level, muscle necrosis | [100] | |
| rabbit | Necrosis and high serum CK levels, myotonia | [103] | |
| Fibrates | |||
| Clofibrate, gemfibrozil | zebrafish | Embryo: induction of embryonic malabsorption syndrome (EMS), very little yolk consumption, small-sized larvae, delayed hatching time, round-shaped neuromuscular junctions, disorganization and less striation of muscular fibres, pericardial oedema, impairing thyroid gland morphogenesis | [119] |
| Clofibric acid | zebrafish | Significant reduction in the growth of a parental generation, decreased triglyceride muscle content, abnormalities in male gonad development with a decrease in the fecundity | [120] |
| Bezafibrate, ciprofibrate | mouse | Limitation of glucose and three-carbon compounds oxidation enhance fatty acid oxidation in liver cells, muscle disorders | [70] |
| Fenofibrate | rat | Increase of CK level, muscle damage, influences on glycolytic enzymes | [101] |
| dog | Skeletal muscle injury, CK elevated level, skeletal muscle fibres necrosis | [78] | |
| Fenofibrate acid | rat | Inhibition of organic anion transporting polypeptide 1B1 (OATP1B1) | [101] |
| Others | |||
| Ezetimibe | zebrafish | Reduction of CK level in HCD-fed zebrafish larvae | [121] |
| Most Recent Methods for Assessing Behavior and Muscle Performance in Zebrafish | ||
|---|---|---|
| Method | Application | Reference |
| Measurement of swimming behavior—mathematical and computational analysis | Quantification of the zebrafish larvae swimming behavior and energetics | [170] |
| Calculating bending moments in swimming fish—experimental data and numerical analysis | Assessment of fish swimming e.g., bending moment pattern, analysis of turning, adult fish swimming at different speeds and accelerations | [172] |
| Measurement of ultrafast zebrafish larval swimming tail muscles contraction—recording and computational analysis | Measurement of the contractile parameters of the muscle in the larval tail in vivo | [171] |
| Cell Tracking Profiler (analysis of muscle stem cell responses to injury)—semi-automated image analysis pipeline, based on cell tracking (3D time-lapse datasets) | Accurate measurement of cell shape and movement | [173] |
| Analysis of stress responses in adult zebrafish—behavioral approach | The analysis of swimming behavior in response to stress, allowing e.g., to examine the pharmacological effects of drugs | [174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubińska-Magiera, M.; Migocka-Patrzałek, M.; Lewandowski, D.; Daczewska, M.; Jagla, K. Zebrafish as a Model for the Study of Lipid-Lowering Drug-Induced Myopathies. Int. J. Mol. Sci. 2021, 22, 5654. https://doi.org/10.3390/ijms22115654
Dubińska-Magiera M, Migocka-Patrzałek M, Lewandowski D, Daczewska M, Jagla K. Zebrafish as a Model for the Study of Lipid-Lowering Drug-Induced Myopathies. International Journal of Molecular Sciences. 2021; 22(11):5654. https://doi.org/10.3390/ijms22115654
Chicago/Turabian StyleDubińska-Magiera, Magda, Marta Migocka-Patrzałek, Damian Lewandowski, Małgorzata Daczewska, and Krzysztof Jagla. 2021. "Zebrafish as a Model for the Study of Lipid-Lowering Drug-Induced Myopathies" International Journal of Molecular Sciences 22, no. 11: 5654. https://doi.org/10.3390/ijms22115654
APA StyleDubińska-Magiera, M., Migocka-Patrzałek, M., Lewandowski, D., Daczewska, M., & Jagla, K. (2021). Zebrafish as a Model for the Study of Lipid-Lowering Drug-Induced Myopathies. International Journal of Molecular Sciences, 22(11), 5654. https://doi.org/10.3390/ijms22115654