
Multi-Omics Model Applied to Cancer Genetics

 , , , and
, , , and
Abstract
:1. Introduction
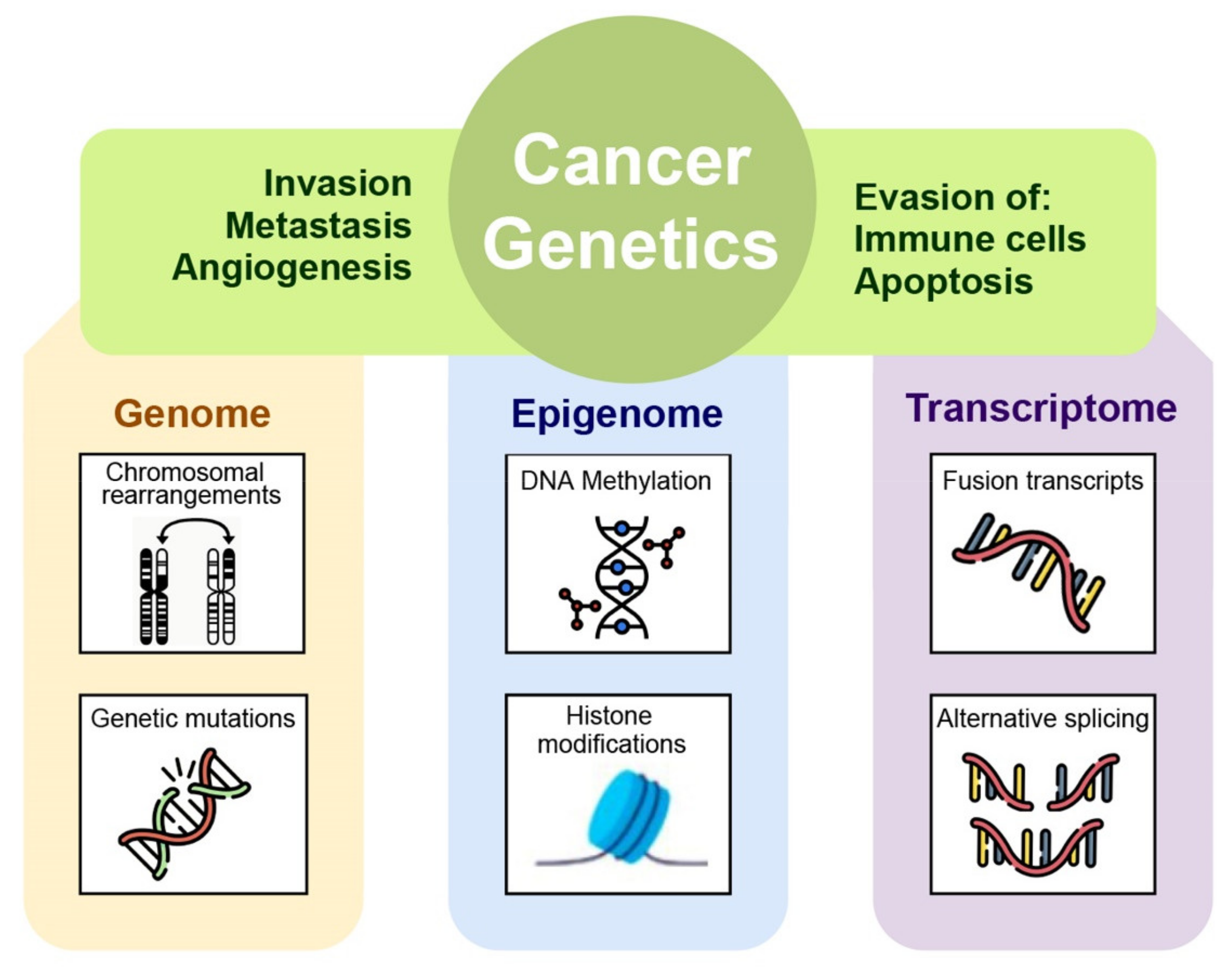
2. Genomics and Molecular Processes
2.1. Cancer Gene Types
- Oncogenes. These, when mutated, actively promote cell proliferation. They are formed when proto-oncogenes that promote cell division are improperly activated, so they are not known to be inherited. They may lead to increased/dysregulated expression of the gene in a new location or to production of fusion proteins with new functions [14]. Two common oncogenes are HER2 and RAS.
- Gatekeeper genes. These are protective genes, also known as tumor suppressor genes. Normally, they negatively control cell growth by monitoring and controlling the cell phases or repairing mismatched DNA.Autosomal recessive mutations in tumor suppressor gene cause loss of function effect at the cellular level, inducing cells to grow uncontrollably, which may eventually form a tumor. Examples of tumor-suppressor genes include BRCA1, BRCA2, and p53 or TP53. Germline mutations in BRCA1 or BRCA2 genes increase a woman’s risk of developing hereditary breast or ovarian cancers and a man’s risk of developing hereditary prostate or breast cancers. They also increase the risk of pancreatic cancer and melanoma in women and men [15]. The most mutated gene in people with cancer is p53 or TP53. More than 50% of cancers involve a missing or damaged p53 gene. Most p53 gene mutations are acquired. Germline p53 mutations are rare, but patients who carry them are at a higher risk of developing many different types of cancer [15].
- Carekeeper genes. These fix the mistakes made when DNA is copied. Many of them function as tumor suppressor genes. BRCA1, BRCA2, and p53 are all DNA repair genes. If a person has an error in a DNA repair gene, mistakes remain uncorrected. Then, the mistakes become mutations. These mutations may eventually lead to cancer, particularly mutations in tumor suppressor genes or oncogenes. Mutations in DNA repair genes may be inherited or acquired. Lynch syndrome is an example of the inherited kind. BRCA1, BRCA2, and p53 mutations and their associated syndromes are also inherited [14].
2.2. Genomic Instability
- Balanced structural changes; the genetic material is equally exchanged, even if genetic information was rearranged into an abnormal gene;
- Unbalanced or nonreciprocal structural changes; the exchange is not equally distributed, and genetic material is added or lost. This can range from the loss or gain of a single base pair to the loss or gain of the entire chromosomes.
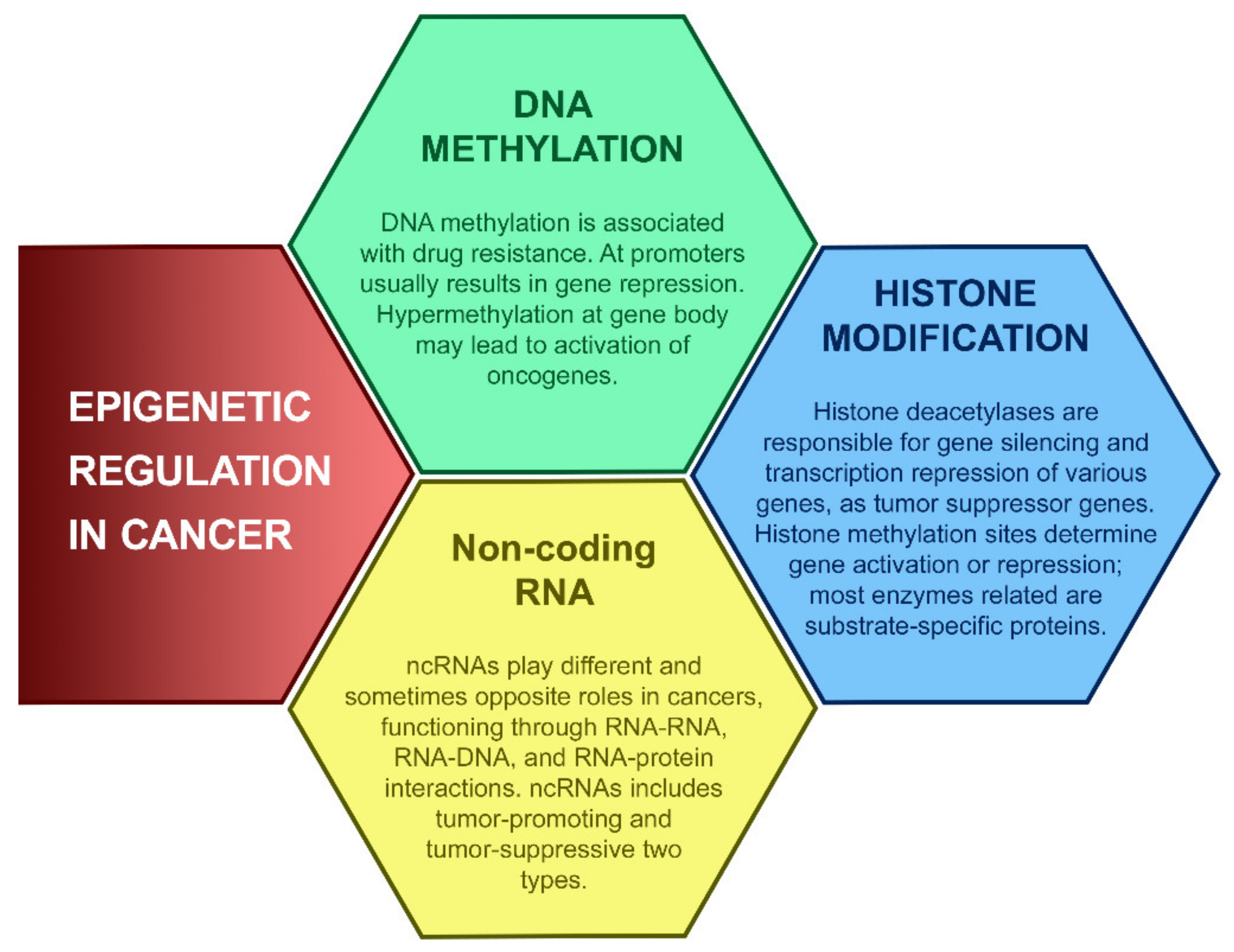
2.3. Epigenomic Instability
3. Roles of Computational Approach in Multi-Omics Era
3.1. Data Acquisition
3.1.1. Genomics
3.1.2. Epigenomics
3.1.3. Transcriptomics
3.1.4. Proteomics and Metabolomics
3.2. Data Management
3.3. Data Integration
3.3.1. Multi-Omics Datasets
- The MultiAssayExperiment Bioconductor database [95] contains the information of different multi-omics experiments, linking features, patients, and experiments;
- The STATegRa dataset [96] has the advantage of allowing the sharing of design principles, increasing their interoperability;
- MOSim tool [97] provides methods for the generation of synthetic multi-omics datasets.
3.3.2. The Problem of Missing Data
3.3.3. Exploratory Data Analysis
3.3.4. Machine Learning Models
3.3.5. Functional Enrichment Approaches
4. Novelty, Challenges, and Future Perspective
- Experimental challenges: an accurate sample preparation in a multi-omics perspective becomes one of the major experimental challenges, with the aim to achieve a universal sample collection and preparation protocol for generating multiple omics datasets.
- Individual omics datasets: data preprocessing is also another significant challenge. This process can be performed on each omic dataset independently before merging significant results or after the production of a unique merged dataset. Moreover, the information included in each individual omic dataset requires very different standardization and scaling approaches, operating in different numerical and time scales.
- Integration issues: data integration issues increases the difficulty of accounting for false positives in merged datasets. Additional problems include the management of rigorous approaches based on statistical models with respect to less rigorous approaches that include a biological interpretation. In comparison to a single omics study, a multi-omics approach has the benefit to allow a deeper understanding of how the tumoral transformation is affecting the flow of information from different omics levels resulting in a bridge between cancerous genotype and the phenotype.
- Data issues: the storage of omics data is very important for reproducibility. To this end, new omic platforms are being developed to provide essential clinical data for insights into the prognosis and diagnosis of diseases.
- Biological knowledge: the interpretation of the outputs of computational models requires a deep knowledge of the biological system under study, in order to discriminate results that are not biologically relevant.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Translocation | Associated Diseases | Fused Genes/Proteins | |
|---|---|---|---|
| First | Second | ||
| t(8;14)(q24;q32) | Burkitt’s lymphoma | c-myc on chromosome 8 | IGH@ (immunoglobulin heavy locus) on chromosome 14 |
| gives the fusion protein lymphocyte-proliferative ability | induces massive transcription of fusion protein | ||
| t(11;14)(q13;q32) | Mantle cell lymphoma | cyclin D1 on chromosome 11 | IGH@ (immunoglobulin heavy locus) on chromosome 14 |
| gives fusion protein cell-proliferative ability | induces massive transcription of fusion protein | ||
| t(14;18)(q32;q21) | Follicular lymphoma (~90% of cases) | IGH@ (immunoglobulin heavy locus) on chromosome 14 | Bcl-2 on chromosome 18 |
| induces massive transcription of fusion protein | gives fusion protein anti-apoptotic abilities | ||
| t(10;(various))(q11;(various)) | Papillary thyroid cancer | RET proto-oncogene on chromosome 10 | PTC (papillary thyroid cancer)—Placeholder for any of several other genes/proteins |
| t(2;3)(q13;p25) | Follicular thyroid cancer | PAX8—paired box gene 8 on chromosome 2 | PPARγ1 (peroxisome proliferator-activated receptor γ 1) on chromosome 3 |
| t(8;21)(q22;q22) | Acute myeloblastic leukemia with maturation | ETO on chromosome 8 | AML1 on chromosome 21 |
| found in ~7% of new cases of AML, carries a favorable prognosis and predicts good response to cytosine arabinoside therapy | |||
| t(9;22)(q34;q11) Philadelphia chromosome | Chronic myelogenous leukemia (CML), acute lymphoblastic leukemia (ALL) | Abl1 gene on chromosome 9 | BCR (“breakpoint cluster region” on chromosome 22 |
| t(15;17)(q22;q21) | Acute promyelocytic leukemia | PML protein on chromosome 15 | RAR-α on chromosome 17 |
| persistent laboratory detection of the PML-RARA transcript is strong predictor of relapse | |||
| t(12;15)(p13;q25) | Acute myeloid leukemia, congenital fibrosarcoma, secretory breast carcinoma, mammary analogue secretory carcinoma of salivary glands, cellular variant of mesoblastic nephroma | TEL on chromosome 12 | TrkC receptor on chromosome 15 |
| t(9;12)(p24;p13) | CML, ALL | JAK on chromosome 9 | TEL on chromosome 12 |
| t(12;16)(q13;p11) | Myxoid liposarcoma | DDIT3 (formerly CHOP) on chromosome 12 | FUS gene on chromosome 16 |
| t(12;21)(p12;q22) | ALL | TEL on chromosome 12 | AML1 on chromosome 21 |
| t(11;18)(q21;q21) | MALT lymphoma | BIRC3 (API-2) | MLT |
| t(1;11)(q42.1;q14.3) | Schizophrenia | ||
| t(2;5)(p23;q35) | Anaplastic large cell lymphoma | ALK | NPM1 |
| t(11;22)(q24;q11.2–12) | Ewing’s sarcoma | FLI1 | EWS |
| t(17;22) | DFSP | Collagen I on chromosome 17 | Platelet derived growth factor B on chromosome 22 |
| t(1;12)(q21;p13) | Acute myelogenous leukemia | ||
| t(X;18)(p11.2;q11.2) | Synovial sarcoma | ||
| t(1;19)(q10;p10) | Oligodendroglioma and oligoastrocytoma | ||
| t(17;19)(q22;p13) | ALL | ||
| t(7,16) (q32–34;p11) or t(11,16) (p11;p11) | Low-grade fibromyxoid sarcoma | FUS | CREB3L2 or CREB3L1 |
References
- Ferlay, J.; Ervik, M.L.F.; Colombet, M.; Mery, L.; Piñeros, M. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2021. [Google Scholar]
- De Anda-Jáuregui, G.; Hernández-Lemus, E. Computational Oncology in the Multi-Omics Era: State of the Art. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Lemus, E.; Reyes-Gopar, H.; Espinal-Enriquez, J.; Ochoa, S. The Many Faces of Gene Regulation in Cancer: A Computational Oncogenomics Outlook. Genes 2019, 10, 865. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Lu, M.; Cheng, T.; Zhan, X.; Zhan, X. Multiomics-Based Signaling Pathway Network Alterations in Human Non-functional Pituitary Adenomas. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Du, W.; Elemento, O. Cancer systems biology: Embracing complexity to develop better anticancer therapeutic strategies. Oncogene 2015, 34, 3215–3225. [Google Scholar] [CrossRef]
- Chakraborty, S.; Hosen, M.I.; Ahmed, M.; Shekhar, H.U. Onco-Multi-OMICS Approach: A New Frontier in Cancer Research. Biomed. Res. Int. 2018, 2018, 9836256. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer Systems Biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef] [Green Version]
- GuhaThakurta, D.; Sheikh, N.A.; Meagher, T.C.; Letarte, S.; Trager, J.B. Applications of systems biology in cancer immunotherapy: From target discovery to biomarkers of clinical outcome. Expert Rev. Clin. Pharmacol. 2013, 6, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.A.; Samuels, Y. Analysis of the genome to personalize therapy for melanoma. Oncogene 2010, 29, 5545–5555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, M.; Honma, K.; Nakagawa, M. Diversity of genome profiles in malignant lymphoma. Cancer Sci. 2010, 101, 573–578. [Google Scholar] [CrossRef]
- Cigudosa, J.C.; Parsa, N.Z.; Louie, D.C.; Filippa, D.A.; Jhanwar, S.C.; Johansson, B.; Mitelman, F.; Chaganti, R.S. Cytogenetic analysis of 363 consecutively ascertained diffuse large B-cell lymphomas. Genes Chromosomes Cancer 1999, 25, 123–133. [Google Scholar] [CrossRef]
- Society, C.C. Genetic Changes and Cancer Risk. Available online: https://www.cancer.ca/en/cancer-information/cancer-101/what-is-cancer/genes-and-cancer/genetic-changes-and-cancer-risk/?region=on (accessed on 30 May 2020).
- (NIH). The Genetics of Cancer. Available online: https://www.cancer.gov/about-cancer/causes-prevention/genetics (accessed on 30 May 2020).
- Chakravarthi, B.V.; Nepal, S.; Varambally, S. Genomic and Epigenomic Alterations in Cancer. Am. J. Pathol. 2016, 186, 1724–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, I. Chromosome Abnormalities and Cancer Cytogenetics. Nat. Educ. 2008, 1, 25–44. [Google Scholar]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.-J.; Liu, Y.-F.; Xu, L.-Z.; Long, Z.-J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.-X.; Pan, Y.-J.; Yan, J.-S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [Green Version]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt’s lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesson, L.B.; Hitchins, M.P.; Ward, R.L. Epimutations and cancer predisposition: Importance and mechanisms. Curr. Opin. Genet. Dev. 2010, 20, 290–298. [Google Scholar] [CrossRef]
- Hassanpour, S.H.; Dehghani, M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Eguchi, S.; Herman, J.G.; Baylin, S.B. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002, 62, 7213–7218. [Google Scholar] [PubMed]
- Ben-Porath, I.; Cedar, H. Epigenetic crosstalk. Mol. Cell 2001, 8, 933–935. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, P.W. Cancer epigenetics. Hum. Mol. Genet. 2005, 14, R65–R76. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Tost, J. DNA Methylation: An Introduction to the Biology and the Disease-Associated Changes of a Promising Biomarker. Mol. Biotechnol. 2010, 44, 71–81. [Google Scholar] [CrossRef]
- Balmain, A.; Gray, J.; Ponder, B. The genetics and genomics of cancer. Nat. Genet. 2003, 33, 238–244. [Google Scholar] [CrossRef]
- Gelli, E.; Pinto, A.M.; Somma, S.; Imperatore, V.; Cannone, M.G.; Hadjistilianou, T.; De Francesco, S.; Galimberti, D.; Curro, A.; Bruttini, M.; et al. Evidence of predisposing epimutation in retinoblastoma. Hum. Mutat 2019, 40, 201–206. [Google Scholar] [CrossRef]
- Costello, J.F.; Frühwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomäki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type–specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: A multicentre, retrospective analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Esteller, M. Lung cancer epigenetics: From knowledge to applications. Semin. Cancer Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, J.K.; Jung, H. Genome-wide methylation patterns predict clinical benefit of immunotherapy in lung cancer. Clin. Epigenetics 2020, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Goltz, D.; Gevensleben, H.; Vogt, T.J.; Dietrich, J.; Golletz, C.; Bootz, F.; Kristiansen, G.; Landsberg, J.; Dietrich, D. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sigin, V.O.; Kalinkin, A.I.; Kuznetsova, E.B.; Simonova, O.A.; Chesnokova, G.G.; Litviakov, N.V.; Slonimskaya, E.M.; Tsyganov, M.M.; Ibragimova, M.K.; Volodin, I.V.; et al. DNA methylation markers panel can improve prediction of response to neoadjuvant chemotherapy in luminal B breast cancer. Sci. Rep. 2020, 10, 9239. [Google Scholar] [CrossRef]
- Yu, D.-H.; Waterland, R.A.; Zhang, P.; Schady, D.; Chen, M.-H.; Guan, Y.; Gadkari, M.; Shen, L. Targeted p16(Ink4a) epimutation causes tumorigenesis and reduces survival in mice. J. Clin. Investig. 2014, 124, 3708–3712. [Google Scholar] [CrossRef]
- Majumder, A.; Syed, K.M.; Mukherjee, A.; Lankadasari, M.B.; Azeez, J.M.; Sreeja, S.; Harikumar, K.B.; Pillai, M.R.; Dutta, D. Enhanced expression of histone chaperone APLF associate with breast cancer. Mol. Cancer 2018, 17, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Fullgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.; Li, M.; Choi, K.; Balch, C.; Kim, S.; Nephew, K.P. MicroRNA and mRNA integrated analysis (MMIA): A web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 2009, 37, W356–W362. [Google Scholar] [CrossRef]
- Musilova, K.; Mraz, M. MicroRNAs in B-cell lymphomas: How a complex biology gets more complex. Leukemia 2015, 29, 1004–1017. [Google Scholar] [CrossRef]
- Erdogan, B.; Facey, C.; Qualtieri, J.; Tedesco, J.; Rinker, E.; Isett, R.B.; Tobias, J.; Baldwin, D.A.; Thompson, J.E.; Carroll, M.; et al. Diagnostic microRNAs in myelodysplastic syndrome. Exp. Hematol. 2011, 39, 915–926.e2. [Google Scholar] [CrossRef] [PubMed]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Galaxy, T. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [Green Version]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Peter, A.; Michael, R.C.; Nebojša, T.; Brad, C.; John, C.; Michael, H.; Andrey, K.; Dan, L.; Hervé, M.; Maya, N.; et al. Common Workflow Language, v1.0. 2016. Available online: https://escholarship.org/uc/item/25z538jj (accessed on 30 May 2020).
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaharia, M.; Bolosky, W.J.; Curtis, K.; Fox, A.; Patterson, D.; Shenker, S.; Stoica, I.; Karp, R.M.; Sittler, T. Faster and More Accurate Sequence Alignment with SNAP. arXiv 2011, arXiv:1111.5572. [Google Scholar]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Res 2016, 5, 1281. [Google Scholar] [CrossRef]
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Grossman, R.; Stein, L. PeakRanger: A cloud-enabled peak caller for ChIP-seq data. BMC Bioinform. 2011, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Mahony, S.; Gifford, D.K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput. Biol. 2012, 8, e1002638. [Google Scholar] [CrossRef] [Green Version]
- Harmanci, A.; Rozowsky, J.; Gerstein, M. MUSIC: Identification of enriched regions in ChIP-Seq experiments using a mappability-corrected multiscale signal processing framework. Genome Biol. 2014, 15, 474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, Y.H.; Johnson, T.D.; Rozek, L.S.; Sartor, M.A. PePr: A peak-calling prioritization pipeline to identify consistent or differential peaks from replicated ChIP-Seq data. Bioinformatics 2014, 30, 2568–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Muratani, M.; Rayan, N.A.; Kraus, P.; Lufkin, T.; Ng, H.H.; Prabhakar, S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 2013, 31, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Iwamoto, N.; Shimada, T. Recent advances in mass spectrometry-based approaches for proteomics and biologics: Great contribution for developing therapeutic antibodies. Pharmacol. Ther. 2018, 185, 147–154. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Shruthi, B.S.; Vinodhkumar, P.; Selvamani. Proteomics: A new perspective for cancer. Adv. Biomed. Res. 2016, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Yakkioui, Y.; Temel, Y.; Chevet, E.; Negroni, L. Integrated and Quantitative Proteomics of Human Tumors. Methods Enzym. 2017, 586, 229–246. [Google Scholar] [CrossRef]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, E.G.; Ciborowski, M. Applications of Metabolomics in Cancer Studies. Adv. Exp. Med. Biol 2017, 965, 209–234. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Han, X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotti, F.; Rosa, C.; Cicalini, I.; Sacchetta, P.; Del Boccio, P.; Genovesi, D.; Pieragostino, D. Advances in Lipidomics for Cancer Biomarkers Discovery. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggio, R.; Villas-Boas, S.G.; Ruggiero, K. Metab: An R package for high-throughput analysis of metabolomics data generated by GC-MS. Bioinformatics 2011, 27, 2316–2318. [Google Scholar] [CrossRef] [PubMed]
- Stanstrup, J.; Broeckling, C.D.; Helmus, R.; Hoffmann, N.; Mathe, E.; Naake, T.; Nicolotti, L.; Peters, K.; Rainer, J.; Salek, R.M.; et al. The metaRbolomics Toolbox in Bioconductor and beyond. Metabolites 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.; Molendijk, J.; Hill, M.M. lipidr: A Software Tool for Data Mining and Analysis of Lipidomics Datasets. J. Proteome Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.; van den Berg, L.; van Overveld, P.; Boiten, J.W. Research Data Stewardship for Healthcare Professionals. Fundam. Clin. Data Sci. 2019, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S. Collaboration Generates Most Complete Cancer Genome Map. 2020. Available online: https://news.harvard.edu/gazette/story/2020/02/big-step-toward-identifying-all-cancer-causing-genetic-mutations/ (accessed on 30 May 2020).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louhimo, R.; Hautaniemi, S. CNAmet: An R package for integrating copy number, methylation and expression data. Bioinformatics 2011, 27, 887–888. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.C.; Kim, K.H.; Woo, S.M.; Myung, J.K. Clinical multi-omics strategies for the effective cancer management. J. Proteom. 2018, 188, 97–106. [Google Scholar] [CrossRef]
- Ramos, M.; Schiffer, L.; Re, A.; Azhar, R.; Basunia, A.; Rodriguez, C.; Chan, T.; Chapman, P.; Davis, S.R.; Gomez-Cabrero, D.; et al. Software for the Integration of Multiomics Experiments in Bioconductor. Cancer Res. 2017, 77, e39–e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortia. STATegRa: Classes and methods for multi-omics data integration. R Package Version 1.24.0. 2020. Available online: https://www.bioconductor.org/packages/release/bioc/html/STATegRa.html (accessed on 30 May 2020).
- Martínez-Mira, C.; Conesa, A.; Tarazona, S. MOSim: Multi-Omics Simulation in R. bioRxiv 2018, 421834. [Google Scholar] [CrossRef]
- Gomez-Cabrero, D.; Abugessaisa, I.; Maier, D.; Teschendorff, A.; Merkenschlager, M.; Gisel, A.; Ballestar, E.; Bongcam-Rudloff, E.; Conesa, A.; Tegner, J. Data integration in the era of omics: Current and future challenges. BMC Syst. Biol. 2014, 8 (Suppl. 2), 1. [Google Scholar] [CrossRef] [PubMed]
- Voillet, V.; Besse, P.; Liaubet, L.; San Cristobal, M.; Gonzalez, I. Handling missing rows in multi-omics data integration: Multiple imputation in multiple factor analysis framework. BMC Bioinform. 2016, 17, 402. [Google Scholar] [CrossRef] [Green Version]
- Pigott, T.D. A Review of Methods for Missing Data. Educ. Res. Eval. 2001, 7, 353–383. [Google Scholar] [CrossRef] [Green Version]
- Van Iterson, M.; Cats, D.; Hop, P.; BIOS Consortium; Heijmans, B.T. omicsPrint: Detection of data linkage errors in multiple omics studies. Bioinformatics 2018, 34, 2142–2143. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Zeleznik, O.A.; Thallinger, G.G.; Kuster, B.; Gholami, A.M.; Culhane, A.C. Dimension reduction techniques for the integrative analysis of multi-omics data. Brief. Bioinform 2016, 17, 628–641. [Google Scholar] [CrossRef]
- Brazma, A.; Culhane, A.C. Algorithms for gene expression analysis. In Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Streicher, K.L.; Zhu, W.; Lehmann, K.P.; Georgantas, R.W.; Morehouse, C.A.; Brohawn, P.; Carrasco, R.A.; Xiao, Z.; Tice, D.A.; Higgs, B.W.; et al. A novel oncogenic role for the miRNA-506-514 cluster in initiating melanocyte transformation and promoting melanoma growth. Oncogene 2012, 31, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Biton, A.; Bernard-Pierrot, I.; Lou, Y.; Krucker, C.; Chapeaublanc, E.; Rubio-Pérez, C.; López-Bigas, N.; Kamoun, A.; Neuzillet, Y.; Gestraud, P.; et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014, 9, 1235–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Tripathy, D.S. OMICsPCA: An. R Package for Quantitative Integration and Analysis of Multiple Omics Assays from Heterogeneous Samples. R Package Version 1.5.0. 2019. Available online: https://www.bioconductor.org/packages/release/bioc/html/OMICsPCA.html (accessed on 30 May 2020).
- Xu, T.; Le, T.D.; Liu, L.; Su, N.; Wang, R.; Sun, B.; Colaprico, A.; Bontempi, G.; Li, J. CancerSubtypes: An R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 2017, 33, 3131–3133. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Kuster, B.; Culhane, A.C.; Gholami, A.M. A multivariate approach to the integration of multi-omics datasets. BMC Bioinform. 2014, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Mezhoud, K. bioCancer: Interactive Multi-Omics Cancers Data Visualization and Analysis. R package version 1.16.0. 2020. Available online: http://kmezhoud.github.io/bioCancer (accessed on 30 May 2020).
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S. schex: Hexbin plots for single cell omics data. R package version 1.2.0. Available online: https://github.com/SaskiaFreytag/schex (accessed on 30 May 2020).
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sales, G.; Calura, E.; Cavalieri, D.; Romualdi, C. Graphite—A Bioconductor package to convert pathway topology to gene network. BMC Bioinform. 2012, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Syed-Abdul, S.; Iqbal, U.; Jack Li, Y.C. Predictive Analytics through Machine Learning in the clinical settings. Comput Methods Programs Biomed. 2017, 144, A1–A2. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Le Cao, K.A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Rinaudo, P.; Boudah, S.; Junot, C.; Thevenot, E.A. biosigner: A New Method for the Discovery of Significant Molecular Signatures from Omics Data. Front. Mol. Biosci. 2016, 3, 26. [Google Scholar] [CrossRef]
- Hernandez-Ferrer, C.; Wellenius, G.A.; Tamayo, I.; Basagana, X.; Sunyer, J.; Vrijheid, M.; Gonzalez, J.R. Comprehensive study of the exposome and omic data using rexposome Bioconductor Packages. Bioinformatics 2019, 35, 5344–5345. [Google Scholar] [CrossRef]
- Metwally, A.A.; Zhang, T.; Snyder, M. OmicsLonDA: Omics Longitudinal Differential Analysis. R package version 1.4.0. 2020. Available online: https://github.com/aametwally/OmicsLonDA (accessed on 30 May 2020).
- Calura, E.; Martini, P.; Sales, G.; Beltrame, L.; Chiorino, G.; D’Incalci, M.; Marchini, S.; Romualdi, C. Wiring miRNAs to pathways: A topological approach to integrate miRNA and mRNA expression profiles. Nucleic Acids Res. 2014, 42, e96. [Google Scholar] [CrossRef]
- Wachter, A.; Beissbarth, T. pwOmics: An R package for pathway-based integration of time-series omics data using public database knowledge. Bioinformatics 2015, 31, 3072–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, C.; Basunia, A.; Peters, B.; Gholami, A.M.; Kuster, B.; Culhane, A.C. MOGSA: Integrative Single Sample Gene-set Analysis of Multiple Omics Data. Mol. Cell Proteom. 2019, 18, S153–S168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.C.; Merino, G.A.; Llera, A.S.; Fernandez, E.A. Massive integrative gene set analysis enables functional characterization of breast cancer subtypes. J. Biomed. Inf. 2019, 93, 103157. [Google Scholar] [CrossRef] [PubMed]
- Odom, G.J.; Ban, Y.; Colaprico, A.; Liu, L.; Silva, T.C.; Sun, X.; Pico, A.R.; Zhang, B.; Wang, L.; Chen, X. PathwayPCA: An R/Bioconductor Package for Pathway Based Integrative Analysis of Multi-Omics Data. Proteomics 2020, e1900409. [Google Scholar] [CrossRef]
- Dinalankara, W.; Ke, Q.; Xu, Y.; Ji, L.; Pagane, N.; Lien, A.; Matam, T.; Fertig, E.J.; Price, N.D.; Younes, L.; et al. Digitizing omics profiles by divergence from a baseline. Proc. Natl. Acad. Sci. USA 2018, 115, 4545–4552. [Google Scholar] [CrossRef] [Green Version]
- Zeeberg, B.R.; Feng, W.; Wang, G.; Wang, M.D.; Fojo, A.T.; Sunshine, M.; Narasimhan, S.; Kane, D.W.; Reinhold, W.C.; Lababidi, S.; et al. GoMiner: A resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003, 4, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway Analysis: State of the Art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef] [Green Version]
- Rocha, D.; García, I.A.; González Montoro, A.; Llera, A.; Prato, L.; Girotti, M.R.; Soria, G.; Fernández, E.A. Pan-Cancer Molecular Patterns and Biological Implications Associated with a Tumor-Specific Molecular Signature. Cells 2020, 10, 45. [Google Scholar] [CrossRef]
- Baloni, P.; Dinalankara, W.; Earls, J.C.; Knijnenburg, T.A.; Geman, D.; Marchionni, L.; Price, N.D. Identifying Personalized Metabolic Signatures in Breast Cancer. Metabolites 2020, 11, 20. [Google Scholar] [CrossRef]
- Parkinson, D.R.; Johnson, B.E.; Sledge, G.W. Making personalized cancer medicine a reality: Challenges and opportunities in the development of biomarkers and companion diagnostics. Clin. Cancer Res. 2012, 18, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Misra, B.B.; Langefeld, C.D.; Olivier, M.; Cox, L.A. Integrated Omics: Tools, Advances, and Future Approaches. J. Mol. Endocrinol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Tolios, A.; De Las Rivas, J.; Hovig, E.; Trouillas, P.; Scorilas, A.; Mohr, T. Computational approaches in cancer multidrug resistance research: Identification of potential biomarkers, drug targets and drug-target interactions. Drug Resist. Updat. 2020, 48, 100662. [Google Scholar] [CrossRef]
- Agrawal, P. Artificial Intelligence in Drug Discovery and Development. J. Pharmacovigil. 2018, 6, 80–93. [Google Scholar] [CrossRef]


| OMICS | TYPE | PRINCIPLE | APPLICATION | BIOINFORMATICS TOOLS | |
|---|---|---|---|---|---|
| GENOMICS | Whole exome sequencing | NGS | Exome-wide mutational/analysis | BWA Bowtie Bowtie2 SNAP SAM BAM | |
| Whole genome sequencing | NGS | Genome-wide mutational/analysis | |||
| Targeted gene/exome sequencing | Sanger sequencing | Mutational analysis in targeted gene/exon | |||
| EPIGENOMICS | Methylomics | Whole genome bisulfite sequencing | Genome-wide mapping of DNA methylation pattern | Methylation-Array-Analysis SICER2 PeakRanger GEM MUSIC PePr DFilter MACS | |
| ChIP-sequencing | NGS | Genome-wide mapping of epigenetic marks | |||
| TRANSCRIPTOMICS | RNA-sequencing | NGS | Genome-wide differential gene expression analysis | Bowtie STAR kallisto Salmon | |
| Microarray | Hybridization | Differential gene expression analysis | |||
| PROTEOMICS | Deep-proteomics | Mass-spectrometry | Differential protein expression analysis | MaxQuant Perseus | |
| METABOLOMICS | Deep-metabolomics | Mass-spectrometry | Differential metabolite expression analysis | Metab metaRbolomics Lipidr | |
| Package Tools | Description |
|---|---|
| OMICsPCA | Omics-oriented tools for PCA analysis [106] |
| CancerSubtypes | Contains clustering methods for the identification of cancer subpopulations from multi-omics data [107] |
| Omicade4 | Implementation of multiple co-inertia analysis (MCIA) [108] |
| Biocancer | Interactive multi-omics data exploratory instrument [109] |
| iClusterPlus | Integrative cluster analysis combining different types of genomic data [110] |
| Package Tools | Description |
|---|---|
| mixOmics | R package for the multivariate analysis of biological datasets with a specific focus on data exploration, dimension reduction, and visualization [116]. |
| DIABLO | Package for the identification of multi-omic biomarker panels capable of discriminating between multiple phenotypic groups. It can be used to understand the molecular mechanisms that guide a disease [117]. |
| MOFA | Package for discovering the principal sources of variation in multi-omics data sets [118]. |
| Biosigner | Package for the identification of molecular signatures from large omics datasets in the process of developing new diagnostics [119]. |
| omicRexposome | Package that uses high-dimensional exposome data in disease association studies, including its integration with a variety of high-performance data types [120]. |
| OmicsLonDA | Package that identifies the time intervals in which omics functions are significantly different between groups [121]. |
| Micrographite | Package that provides a method to integrate micro-RNA and mRNA data through their association to canonical pathways [122]. |
| pwOmics | Package for integrating multi-omics data, adapted for the study of time series analyses [123]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pettini, F.; Visibelli, A.; Cicaloni, V.; Iovinelli, D.; Spiga, O. Multi-Omics Model Applied to Cancer Genetics. Int. J. Mol. Sci. 2021, 22, 5751. https://doi.org/10.3390/ijms22115751
Pettini F, Visibelli A, Cicaloni V, Iovinelli D, Spiga O. Multi-Omics Model Applied to Cancer Genetics. International Journal of Molecular Sciences. 2021; 22(11):5751. https://doi.org/10.3390/ijms22115751
Chicago/Turabian StylePettini, Francesco, Anna Visibelli, Vittoria Cicaloni, Daniele Iovinelli, and Ottavia Spiga. 2021. "Multi-Omics Model Applied to Cancer Genetics" International Journal of Molecular Sciences 22, no. 11: 5751. https://doi.org/10.3390/ijms22115751
APA StylePettini, F., Visibelli, A., Cicaloni, V., Iovinelli, D., & Spiga, O. (2021). Multi-Omics Model Applied to Cancer Genetics. International Journal of Molecular Sciences, 22(11), 5751. https://doi.org/10.3390/ijms22115751