
Inferring Single-Cell 3D Chromosomal Structures Based on the Lennard-Jones Potential

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Evaluations
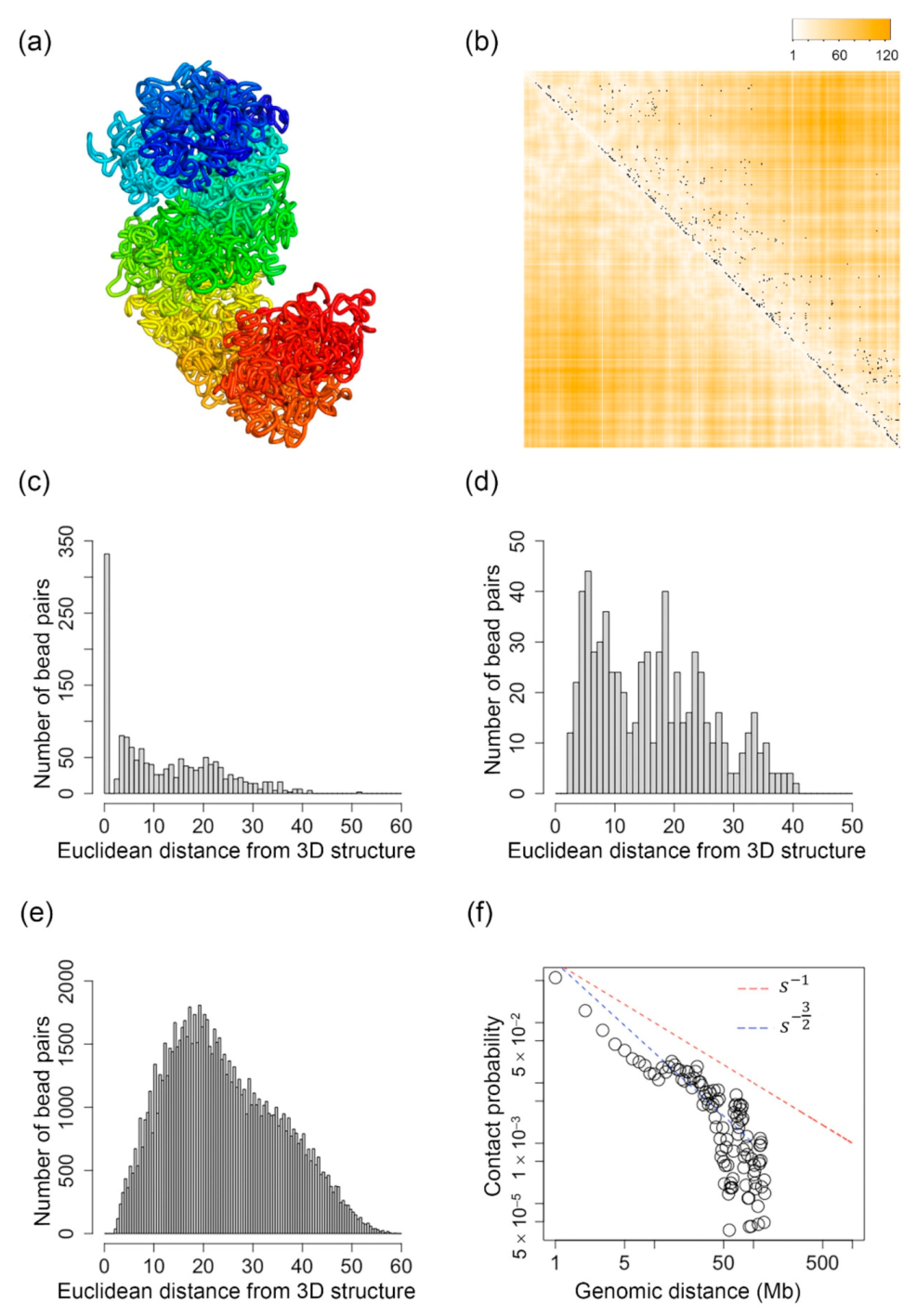
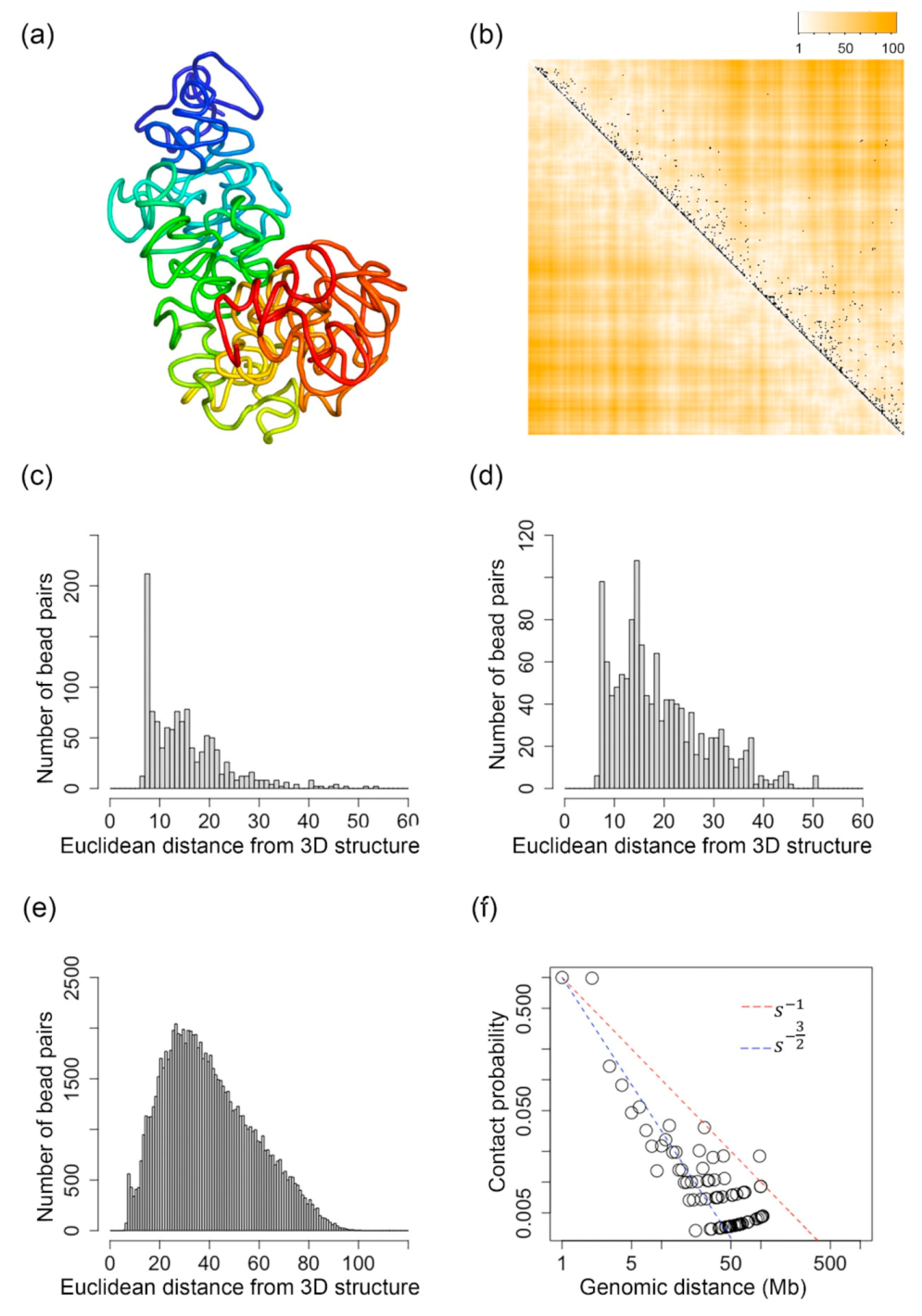
2.1. Inferred 3D Structures of the X-Chromosome of a Mouse TH1 Cell
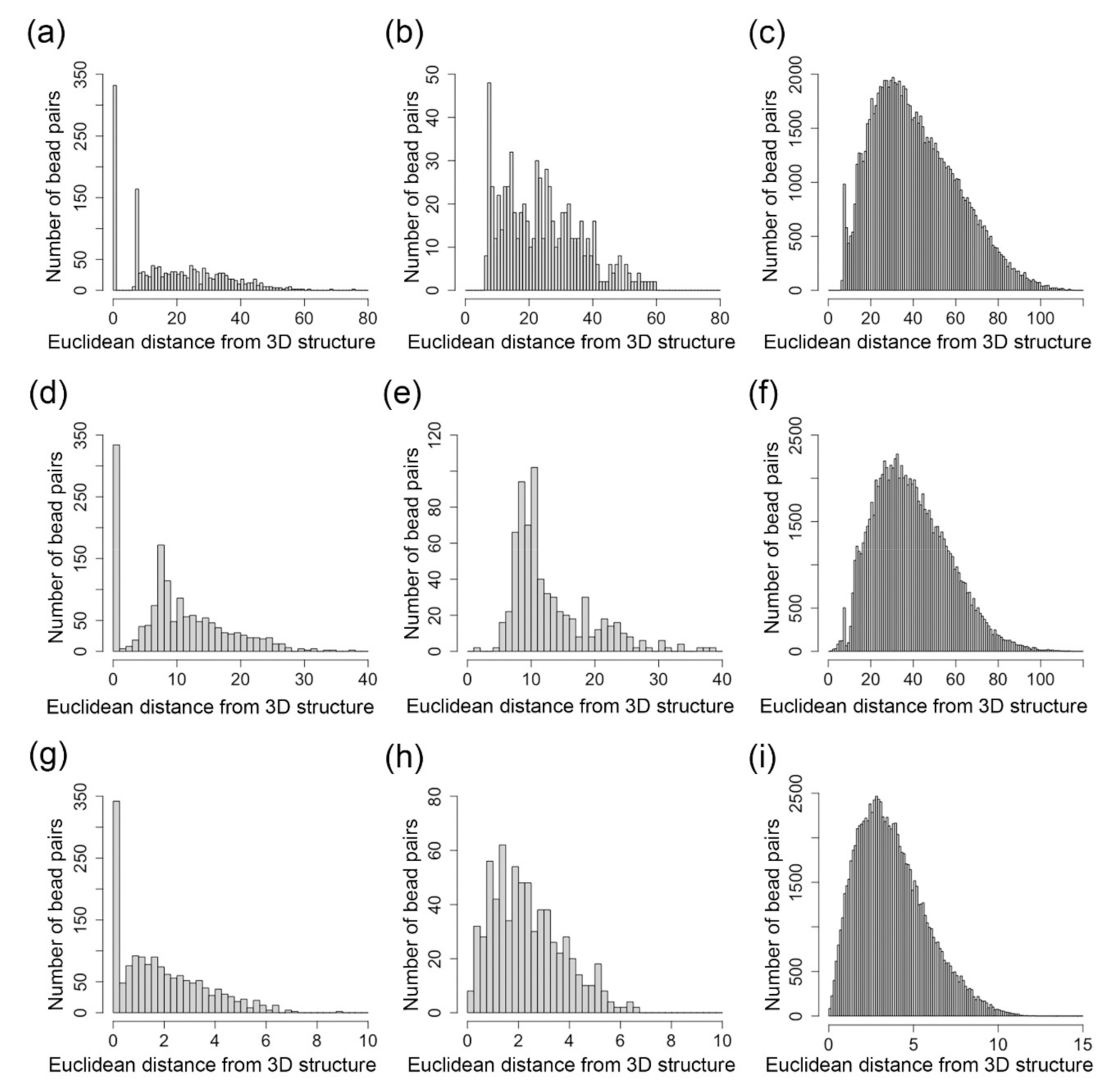
2.2. Pearson’s Correlation Between and Euclidean Distances Parsed from the Inferred 3D Structures
2.3. Comparison with Existing Tools
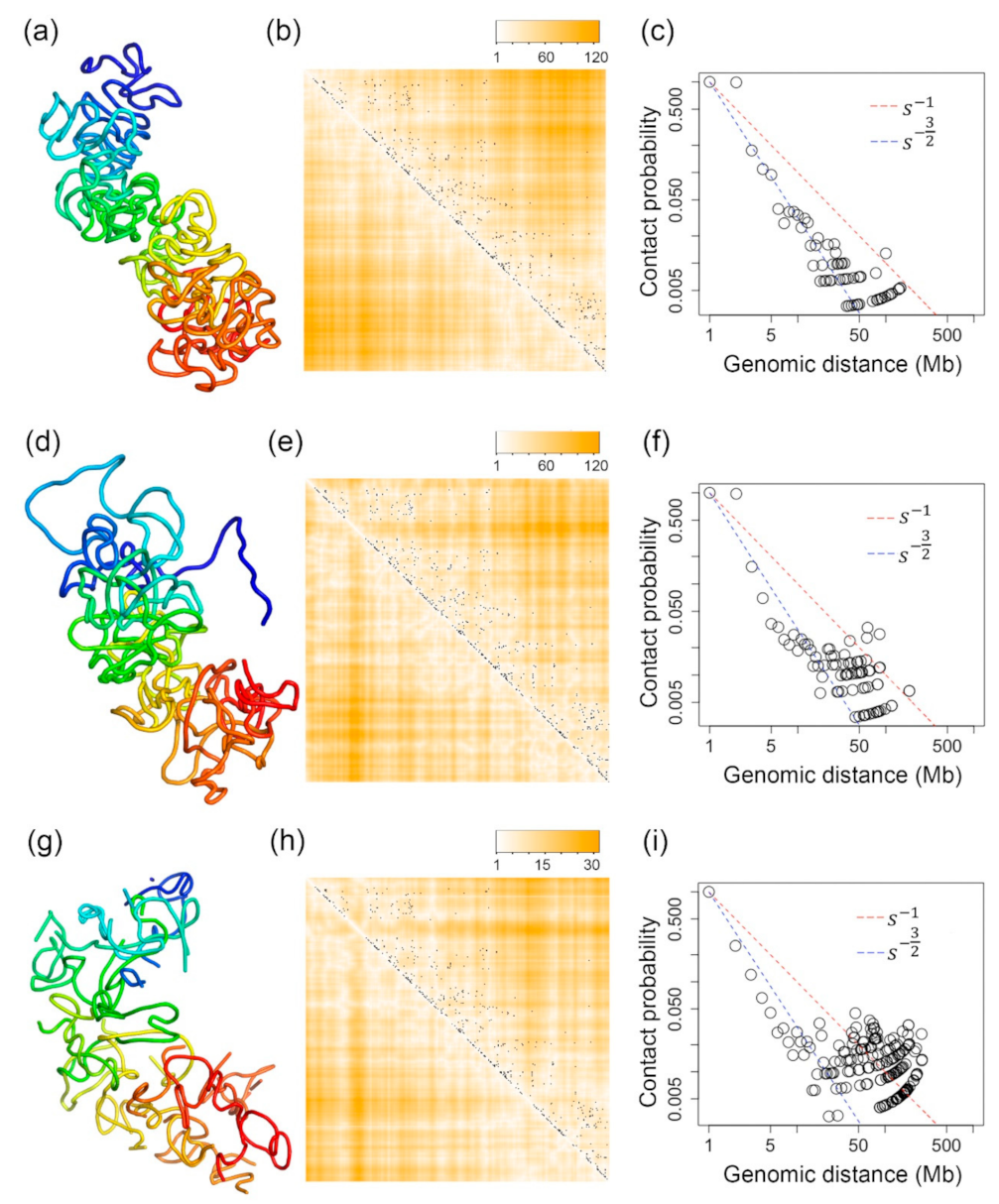
2.4. Inferred 3D Structures of the Active and Inactive X-Chromosomes of a Human GM12878 Cell
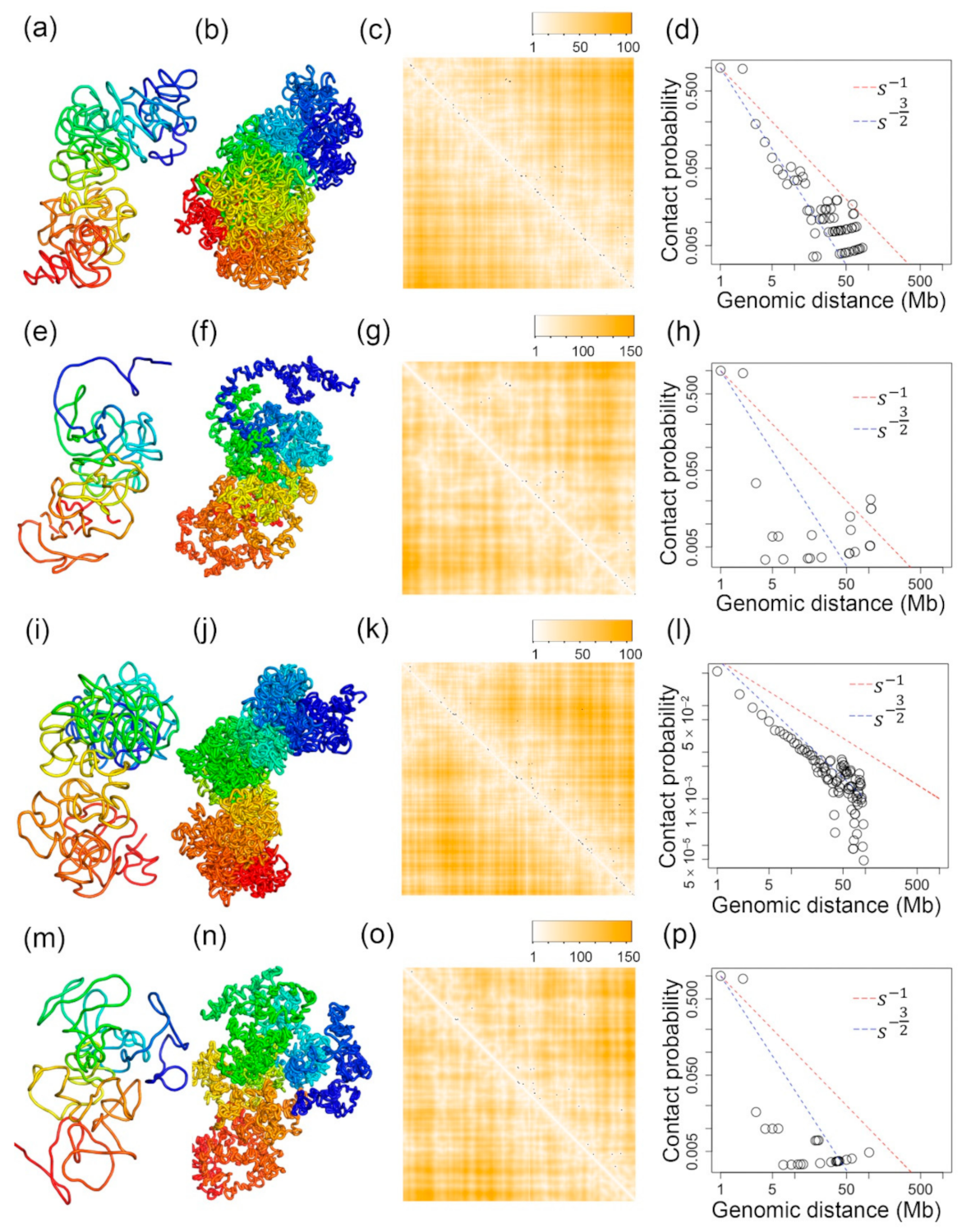
2.5. Inferred 3D Structures of the Chromosome 3 of a Mouse Oocyte Cell
2.6. Validation with 3D-FISH
3. Discussion
4. Materials and Methods
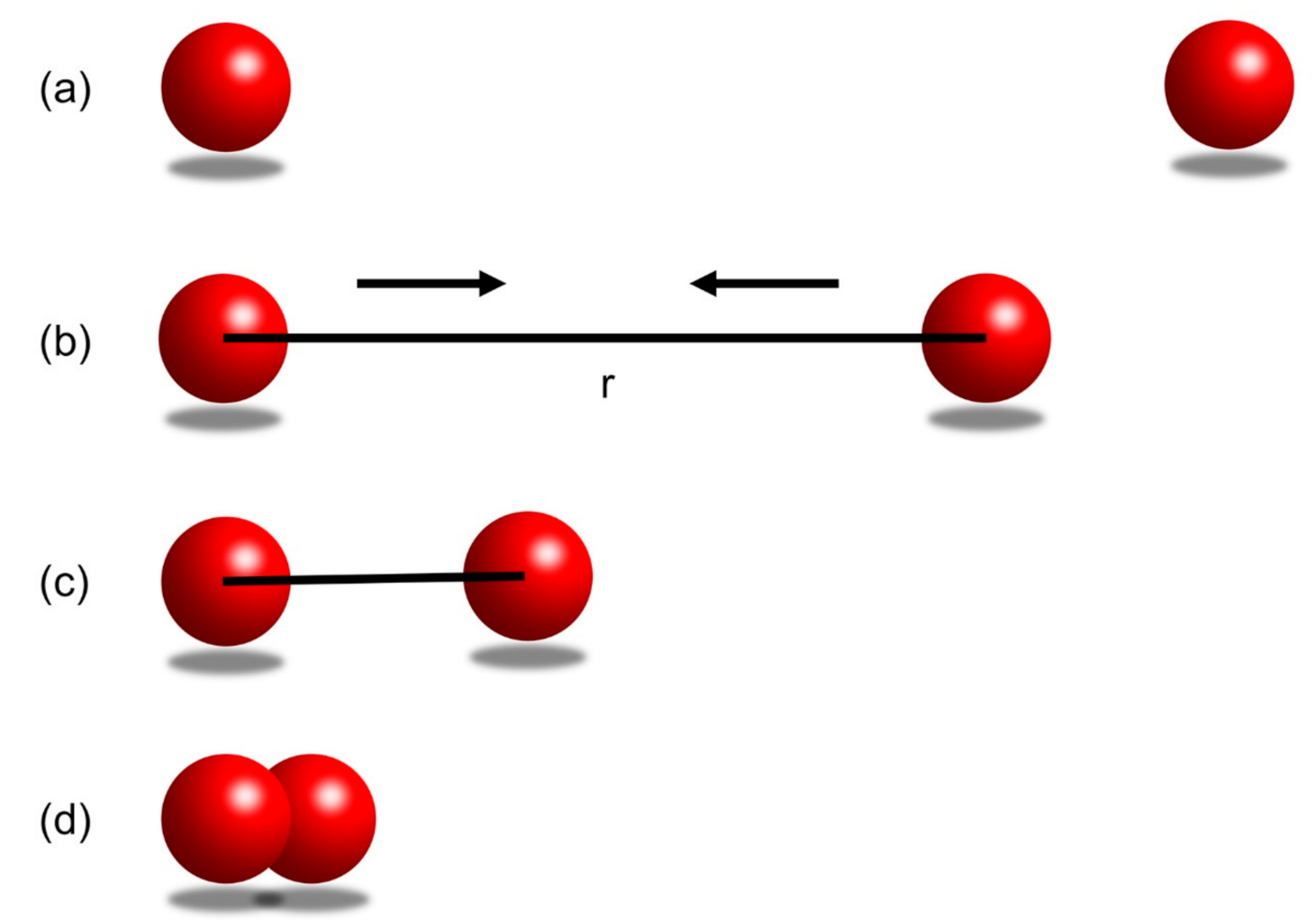
4.1. Introduction to Negative Potential Energy
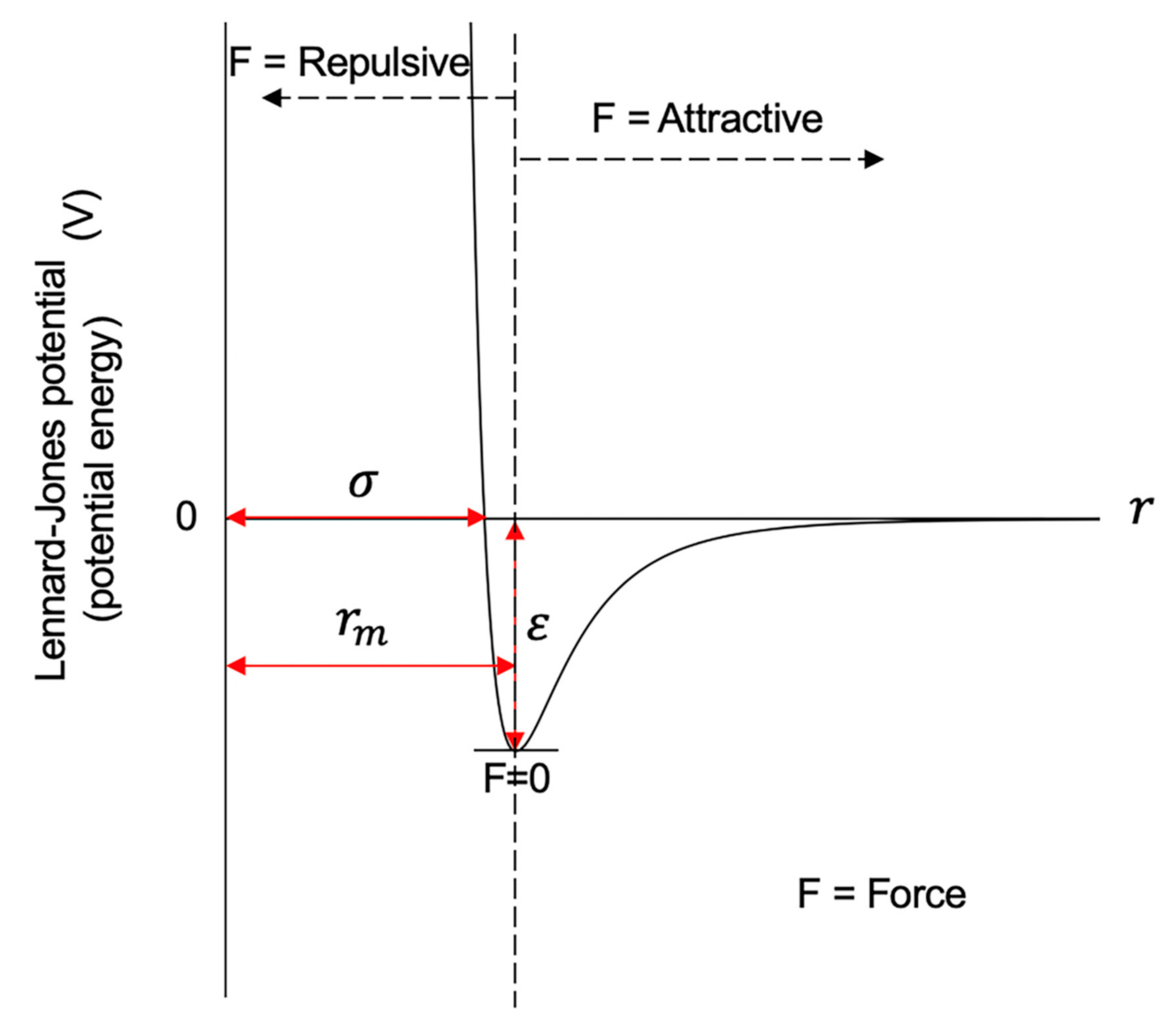
4.2. The Lennard-Jones Potential
4.3. 2D Gaussian Function for Imputing Single-Cell Hi-C Contacts
4.4. Loss Function
4.5. Initialization of Random 3D Chromosomal Structures
4.6. The Metropolis–Hastings Simulations
4.7. Model Selection
4.8. Computational Time
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bickmore, W.A.; van Steensel, B. Genome architecture: Domain organization of interphase chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef] [Green Version]
- Sexton, T.; Cavalli, G. The role of chromosome domains in shaping the functional genome. Cell 2015, 160, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pombo, A.; Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015, 16, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Therizols, P.; Illingworth, R.S.; Courilleau, C.; Boyle, S.; Wood, A.J.; Bickmore, W.A. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 2014, 346, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Towbin, B.D.; Kalck, V.; Cabianca, D.S.; Gaidatzis, D.; Hauer, M.H.; Geng, L.; Wang, L.; Yang, T.; Wang, X. Perinuclear anchoring of H3K9-methylated chromatin stabilizes induced cell fate in C. elegans embryos. Cell 2015, 163, 1333–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wang, Z. Reconstructing high-resolution chromosome three-dimensional structures by hi-C complex networks. BMC Bioinform. 2018, 19, 39–50. [Google Scholar] [CrossRef]
- Liu, T.; Porter, J.; Zhao, C.; Zhu, H.; Wang, N.; Sun, Z.; Mo, Y.-Y.; Wang, Z. TADKB: Family classification and a knowledge base of topologically associating domains. BMC Genom. 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; De Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Miele, A.; Dekker, J. Mapping cis-and trans-chromatin interaction networks using chromosome conformation capture (3C). In The Nucleus; Springer: Berlin/Heidelberg, Germany, 2008; pp. 105–121. [Google Scholar]
- Rodley, C.; Bertels, F.; Jones, B.; O’sullivan, J. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genet. Biol. 2009, 46, 879–886. [Google Scholar] [CrossRef]
- Hagège, H.; Klous, P.; Braem, C.; Splinter, E.; Dekker, J.; Cathala, G.; De Laat, W.; Forné, T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2007, 2, 1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Splinter, E.; Grosveld, F.; de Laat, W. 3C technology: Analyzing the spatial organization of genomic loci in vivo. In Methods in Enzymology; Elsevier: Berlin/Heidelberg, Germany, 2003; Volume 375, pp. 493–507. [Google Scholar]
- De Laat, W.; Dekker, J. 3C-based technologies to study the shape of the genome. Methods 2012, 58. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Lu, C. Microfluidics-based chromosome conformation capture (3C) technology for examining chromatin organization with a low quantity of cells. Anal. Chem. 2018, 90, 3714–3719. [Google Scholar] [CrossRef] [PubMed]
- Hövel, I.; Louwers, M.; Stam, M. 3C technologies in plants. Methods 2012, 58, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Zhao, K. 3C-based methods to detect long-range chromatin interactions. Front. Biol. 2011, 6, 76–81. [Google Scholar] [CrossRef]
- Louwers, M.; Splinter, E.; Van Driel, R.; De Laat, W.; Stam, M. Studying physical chromatin interactions in plants using Chromosome Conformation Capture (3C). Nat. Protoc. 2009, 4, 1216. [Google Scholar] [CrossRef] [Green Version]
- Van de Werken, H.J.; de Vree, P.J.; Splinter, E.; Holwerda, S.J.; Klous, P.; de Wit, E.; de Laat, W. 4C technology: Protocols and data analysis. In Methods in Enzymology; Elsevier: Berlin/Heidelberg, Germany, 2012; Volume 513, pp. 89–112. [Google Scholar]
- Splinter, E.; de Wit, E.; van de Werken, H.J.; Klous, P.; de Laat, W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation. Methods 2012, 58, 221–230. [Google Scholar] [CrossRef]
- Simonis, M.; Klous, P.; Homminga, I.; Galjaard, R.-J.; Rijkers, E.-J.; Grosveld, F.; Meijerink, J.P.; De Laat, W. High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nat. Methods 2009, 6, 837. [Google Scholar] [CrossRef]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; De Wit, E.; Van Steensel, B.; De Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet. 2006, 38, 1348–1354. [Google Scholar] [CrossRef]
- De Laat, W.; Grosveld, F. Capture and Characterized co-Localized Chromatin (4C) Technology. 2012. Available online: https://patents.google.com/patent/US8153373B2/en (accessed on 30 May 2021).
- Simonis, M.; Kooren, J.; De Laat, W. An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods 2007, 4, 895–901. [Google Scholar] [CrossRef]
- Van De Werken, H.J.; Landan, G.; Holwerda, S.J.; Hoichman, M.; Klous, P.; Chachik, R.; Splinter, E.; Valdes-Quezada, C.; Öz, Y.; Bouwman, B.A. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods 2012, 9, 969. [Google Scholar] [CrossRef]
- Gavrilov, A.; Eivazova, E.; Pirozhkova, I.; Lipinski, M.; Razin, S.; Vassetzky, Y. Chromosome conformation capture (from 3C to 5C) and its ChIP-based modification. In Chromatin Immunoprecipitation Assays; Springer: Berlin/Heidelberg, Germany, 2009; pp. 171–188. [Google Scholar]
- Dostie, J.; Dekker, J. Mapping networks of physical interactions between genomic elements using 5C technology. Nat. Protoc. 2007, 2, 988. [Google Scholar] [CrossRef]
- Van Berkum, N.L.; Dekker, J. Determining spatial chromatin organization of large genomic regions using 5C technology. In Chromatin Immunoprecipitation Assays; Springer: Berlin/Heidelberg, Germany, 2009; pp. 189–213. [Google Scholar]
- Ferraiuolo, M.A.; Sanyal, A.; Naumova, N.; Dekker, J.; Dostie, J. From cells to chromatin: Capturing snapshots of genome organization with 5C technology. Methods 2012, 58, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraiuolo, M.A.; Sanyal, A.; Naumova, N.; Dekker, J.; Dostie, J. Mapping chromatin interactions with 5C technology: 5C; a quantitative approach to capturing chromatin conformation over large genomic distances. Methods 2012, 58. [Google Scholar] [CrossRef] [Green Version]
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. JoVE 2010, 39, e1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy-Leduc, C.; Delattre, M.; Mary-Huard, T.; Robin, S. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics 2014, 30, i386–i392. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhang, F.; Yardımcı, G.G.; Song, F.; Hardison, R.C.; Noble, W.S.; Yue, F.; Li, Q. HiCRep: Assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017, 27, 1939–1949. [Google Scholar] [CrossRef] [Green Version]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Serra, F.; Baù, D.; Goodstadt, M.; Castillo, D.; Filion, G.; Marti-Renom, M.A. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput. Biol. 2017, 13, e1005665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; An, L.; Xu, J.; Zhang, B.; Zheng, W.J.; Hu, M.; Tang, J.; Yue, F. Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Fu, L.-Y.; Dong, P.-F.; Deng, Z.-L.; Li, J.-X.; Wang, X.-T.; Zhang, H.-Y. The sequencing bias relaxed characteristics of Hi-C derived data and implications for chromatin 3D modeling. Nucleic Acids Res. 2013, 41, e183. [Google Scholar] [CrossRef] [Green Version]
- Stansfield, J.C.; Cresswell, K.G.; Vladimirov, V.I.; Dozmorov, M.G. HiCcompare: An R-package for joint normalization and comparison of HI-C datasets. BMC Bioinform. 2018, 19, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Deng, K.; Qin, Z.; Liu, J.S. Understanding spatial organizations of chromosomes via statistical analysis of Hi-C data. Quant. Biol. 2013, 1, 156–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Deng, K.; Selvaraj, S.; Qin, Z.; Ren, B.; Liu, J.S. HiCNorm: Removing biases in Hi-C data via Poisson regression. Bioinformatics 2012, 28, 3131–3133. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Hong, P.; Zhang, S.; Xu, W.; Jamal, M.; Yan, K.; Lei, Y.; Li, L.; Ruan, Y.; Fu, Z.F. Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture. Nat. Genet. 2018, 50, 754–763. [Google Scholar] [CrossRef]
- Wu, H.-J.; Michor, F. A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 2016, 32, 3695–3701. [Google Scholar] [CrossRef]
- Ning, C.; He, M.; Tang, Q.; Zhu, Q.; Li, M.; Li, D. Advances in mammalian three-dimensional genome by using Hi-C technology approach. Yi Chuan Hered. 2019, 41, 215–233. [Google Scholar]
- Battulin, N.; Fishman, V.S.; Mazur, A.M.; Pomaznoy, M.; Khabarova, A.A.; Afonnikov, D.A.; Prokhortchouk, E.B.; Serov, O.L. Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 2015, 16, 77. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.; Williamson, I.; Bickmore, W.A.; Dostie, J. An overview of genome organization and how we got there: From FISH to Hi-C. Microbiol. Mol. Biol. Rev. 2015, 79, 347–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, G.; Toh, K.-C.; Sung, W.-K. 3D chromosome modeling with semi-definite programming and Hi-C data. J. Comput. Biol. 2013, 20, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Belaghzal, H.; Dekker, J.; Gibcus, J.H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 2017, 123, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Z. HiCNN: A very deep convolutional neural network to better enhance the resolution of Hi-C data. Bioinformatics 2019, 35, 4222–4228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandão, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Q.; Li, N.; Li, X.; Yuan, Z.; Xie, D.; Wang, X.; Li, J.; Yu, Y.; Wang, J.; Ding, B. Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice. Plant J. 2018, 94, 1141–1156. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, W.; Li, X. 3D genome reconstruction with ShRec3D+ and Hi-C data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 15, 460–468. [Google Scholar] [CrossRef]
- Noble, W.; Duan, Z.-j.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A. A Three-Dimensional Model of the Yeast Genome. In Proceedings of the International Conference on Research in Computational Molecular Biology, Vancouver, BC, Canada, 28–31 March 2011; Springer: Berlin/Heidelberg, Germany, 2011; p. 320. [Google Scholar]
- Baù, D.; Marti-Renom, M.A. Genome structure determination via 3C-based data integration by the Integrative Modeling Platform. Methods 2012, 58, 300–306. [Google Scholar] [CrossRef]
- Rousseau, M.; Fraser, J.; Ferraiuolo, M.A.; Dostie, J.; Blanchette, M. Three-dimensional modeling of chromatin structure from interaction frequency data using Markov chain Monte Carlo sampling. BMC Bioinform. 2011, 12, 414. [Google Scholar] [CrossRef] [Green Version]
- Trussart, M.; Serra, F.; Baù, D.; Junier, I.; Serrano, L.; Marti-Renom, M.A. Assessing the limits of restraint-based 3D modeling of genomes and genomic domains. Nucleic Acids Res. 2015, 43, 3465–3477. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, G.; Toh, K.-C.; Sung, W.-K. Inference of Spatial Organizations of Chromosomes Using Semi-Definite Embedding Approach and Hi-C Data. In Proceedings of the Annual International Conference on Research in Computational Molecular Biology, Padua, Italy, 10–13 May 2013; Springer: Berlin/Heidelberg, Germany, 2013; pp. 317–332. [Google Scholar]
- Hu, M.; Deng, K.; Qin, Z.; Dixon, J.; Selvaraj, S.; Fang, J.; Ren, B.; Liu, J.S. Bayesian inference of spatial organizations of chromosomes. PLoS Comput. Biol. 2013, 9, e1002893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varoquaux, N.; Ay, F.; Noble, W.S.; Vert, J.-P. A statistical approach for inferring the 3D structure of the genome. Bioinformatics 2014, 30, i26–i33. [Google Scholar] [CrossRef] [PubMed]
- Trieu, T.; Cheng, J. MOGEN: A tool for reconstructing 3D models of genomes from chromosomal conformation capturing data. Bioinformatics 2016, 32, 1286–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieber, L.; Mahony, S. miniMDS: 3D structural inference from high-resolution Hi-C data. Bioinformatics 2017, 33, i261–i266. [Google Scholar] [CrossRef] [Green Version]
- Wlasnowolski, M.; Sadowski, M.; Czarnota, T.; Jodkowska, K.; Szalaj, P.; Tang, Z.; Ruan, Y.; Plewczynski, D. 3D-GNOME 2.0: A three-dimensional genome modeling engine for predicting structural variation-driven alterations of chromatin spatial structure in the human genome. Nucleic Acids Res. 2020, 48, W170–W176. [Google Scholar] [CrossRef]
- Zhu, G.; Deng, W.; Hu, H.; Ma, R.; Zhang, S.; Yang, J.; Peng, J.; Kaplan, T.; Zeng, J. Reconstructing spatial organizations of chromosomes through manifold learning. Nucleic Acids Res. 2018, 46, e50. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.; He, X.; Niu, J.; Zhou, B.; Zhu, G.; Ma, T.; Song, J.; Gao, J.; Zhang, M.Q.; Zeng, J. Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudai, C.; Salerno, E.; Zoppe, M.; Merelli, I.; Tonazzini, A. ChromStruct 4: A python code to estimate the chromatin structure from hi-C data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Hua, N.; Tjong, H.; Shin, H.; Gong, K.; Zhou, X.J.; Alber, F. Producing genome structure populations with the dynamic and automated PGS software. Nat. Protoc. 2018, 13, 915. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Bonev, B.; Cohen, N.M.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A. Multiscale 3D genome rewiring during mouse neural development. Cell 2017, 171, 557–572.e24. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Várnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Cohen, N.M.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Xing, D.; Chang, C.-H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Carstens, S.; Nilges, M.; Habeck, M. Inferential structure determination of chromosomes from single-cell Hi-C data. PLoS Comput. Biol. 2016, 12, e1005292. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, M.; Bryner, D.; Huffer, F.; Evans, S.; Srivastava, A.; Neretti, N. Bayesian estimation of three-dimensional chromosomal structure from single-cell hi-c data. J. Comput. Biol. 2019, 26, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, Z. SCL: A lattice-based approach to infer 3D chromosome structures from single-cell Hi-C data. Bioinformatics 2019, 35, 3981–3988. [Google Scholar] [CrossRef] [Green Version]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Sanborn, A.L.; Rao, S.S.; Huang, S.-C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.-M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, M.; Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Esposito, A.; Nicodemi, M. Polymer physics indicates chromatin folding variability across single-cells results from state degeneracy in phase separation. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Jost, D.; Carrivain, P.; Cavalli, G.; Vaillant, C. Modeling epigenome folding: Formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014, 42, 9553–9561. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, M.; Zhang, B.; Aiden, E.L.; Wolynes, P.G.; Onuchic, J.N. Transferable model for chromosome architecture. Proc. Natl. Acad. Sci. USA 2016, 113, 12168–12173. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Polycarpou, A.A. Adhesive contact based on the Lennard–Jones potential: A correction to the value of the equilibrium distance as used in the potential. J. Colloid Interface Sci. 2004, 278, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Semiromi, D.T.; Azimian, A. Molecular dynamics simulation of nonodroplets with the modified Lennard-Jones potential function. Heat Mass Transf. 2011, 47, 579–588. [Google Scholar] [CrossRef]
- Smit, B. Phase diagrams of Lennard-Jones fluids. J. Chem. Phys. 1992, 96, 8639–8640. [Google Scholar] [CrossRef] [Green Version]
- Mastny, E.A.; de Pablo, J.J. Melting line of the Lennard-Jones system, infinite size, and full potential. J. Chem. Phys. 2007, 127, 104504. [Google Scholar] [CrossRef]
- Wei-Zhong, L.; Cong, C.; Jian, Y. Molecular dynamics simulation of self-diffusion coefficient and its relation with temperature using simple Lennard-Jones potential. Heat Transf. 2008, 37, 86–93. [Google Scholar] [CrossRef]
- Pandey, R.B.; Farmer, B.L. Conformational response to solvent interaction and temperature of a protein (histone h3. 1) by a multi-grained Monte Carlo simulation. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A.; Noble, W.S. A three-dimensional model of the yeast genome. Nature 2010, 465, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; de Santiago, I.; Lavitas, L.-M.; Branco, M.R. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 2017, 543, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Binder, K. Monte Carlo and Molecular Dynamics Simulations in Polymer Science; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by simulated annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zha, M.; Wang, N.; Zhang, C.; Wang, Z. Inferring Single-Cell 3D Chromosomal Structures Based on the Lennard-Jones Potential. Int. J. Mol. Sci. 2021, 22, 5914. https://doi.org/10.3390/ijms22115914
Zha M, Wang N, Zhang C, Wang Z. Inferring Single-Cell 3D Chromosomal Structures Based on the Lennard-Jones Potential. International Journal of Molecular Sciences. 2021; 22(11):5914. https://doi.org/10.3390/ijms22115914
Chicago/Turabian StyleZha, Mengsheng, Nan Wang, Chaoyang Zhang, and Zheng Wang. 2021. "Inferring Single-Cell 3D Chromosomal Structures Based on the Lennard-Jones Potential" International Journal of Molecular Sciences 22, no. 11: 5914. https://doi.org/10.3390/ijms22115914
APA StyleZha, M., Wang, N., Zhang, C., & Wang, Z. (2021). Inferring Single-Cell 3D Chromosomal Structures Based on the Lennard-Jones Potential. International Journal of Molecular Sciences, 22(11), 5914. https://doi.org/10.3390/ijms22115914