
Protein Structure Refinement Using Multi-Objective Particle Swarm Optimization with Decomposition Strategy


Abstract
:1. Introduction
2. Experiments and Results
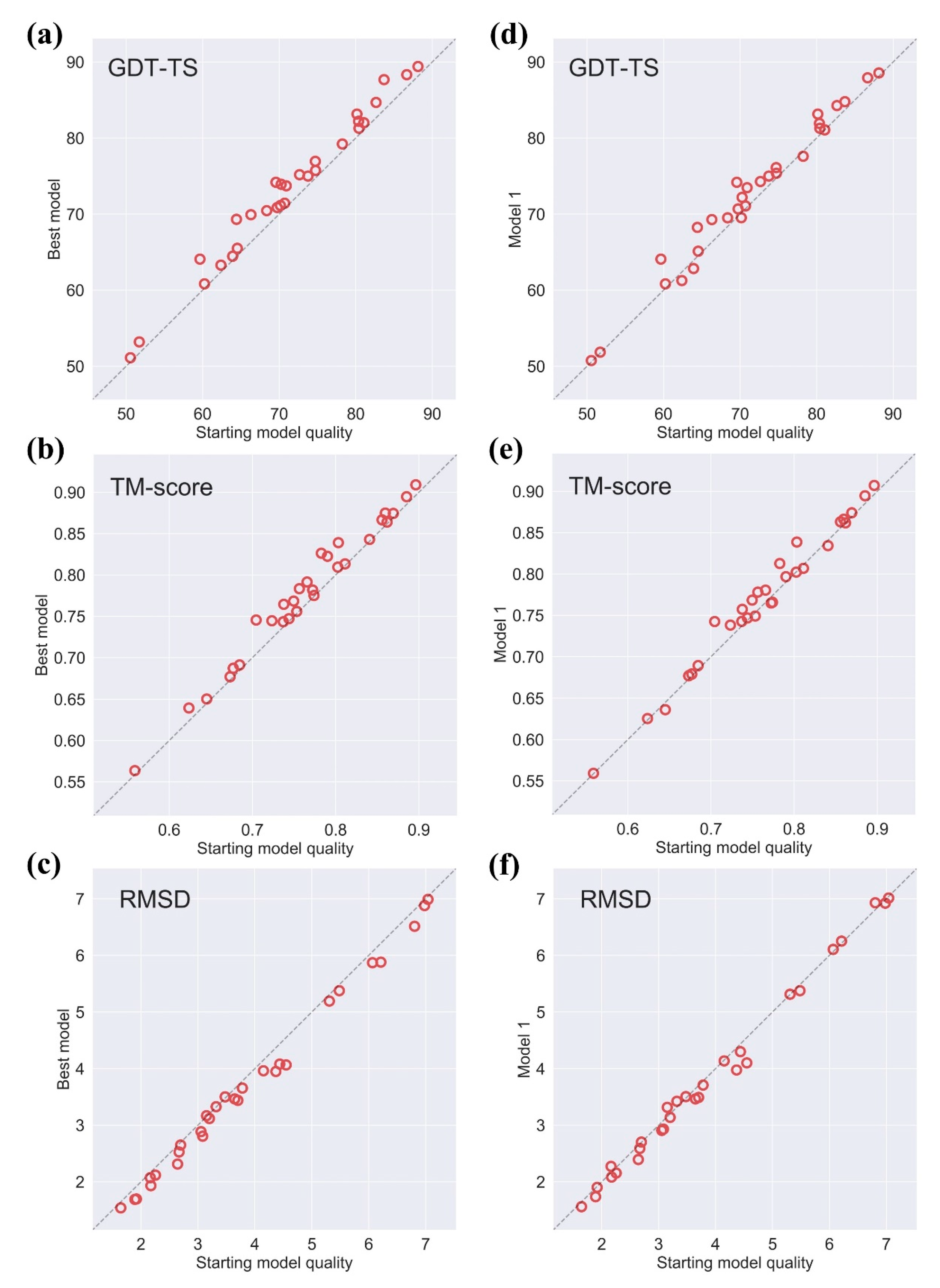
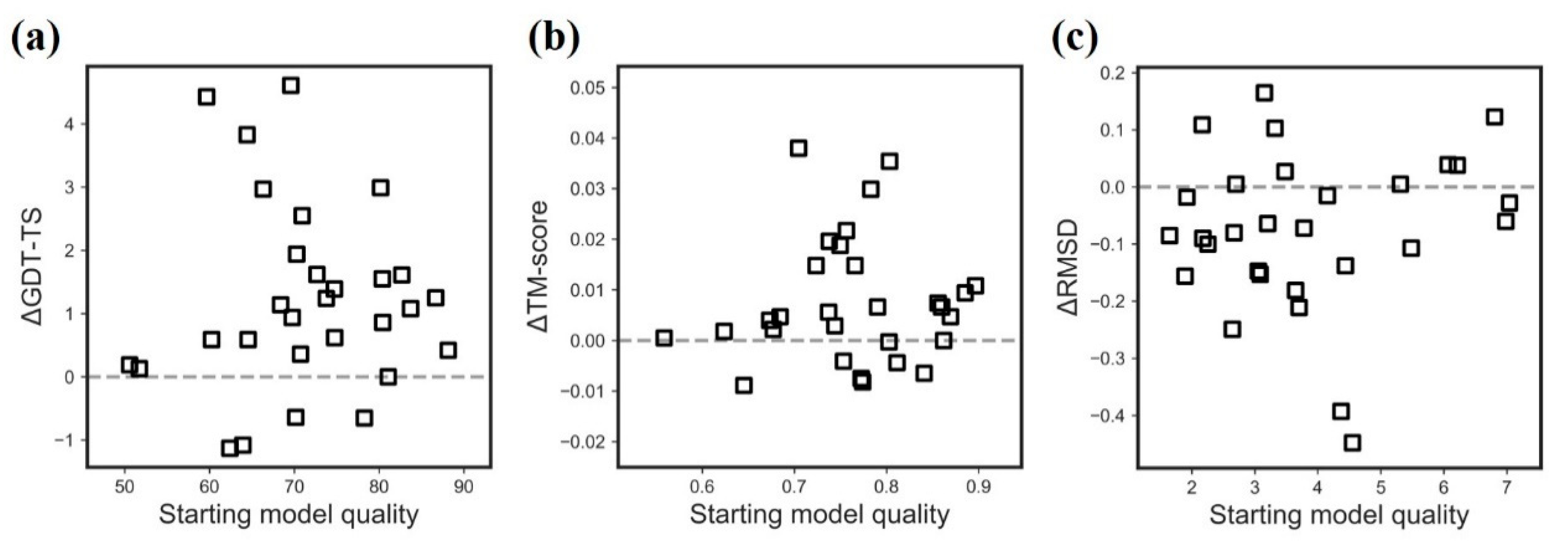
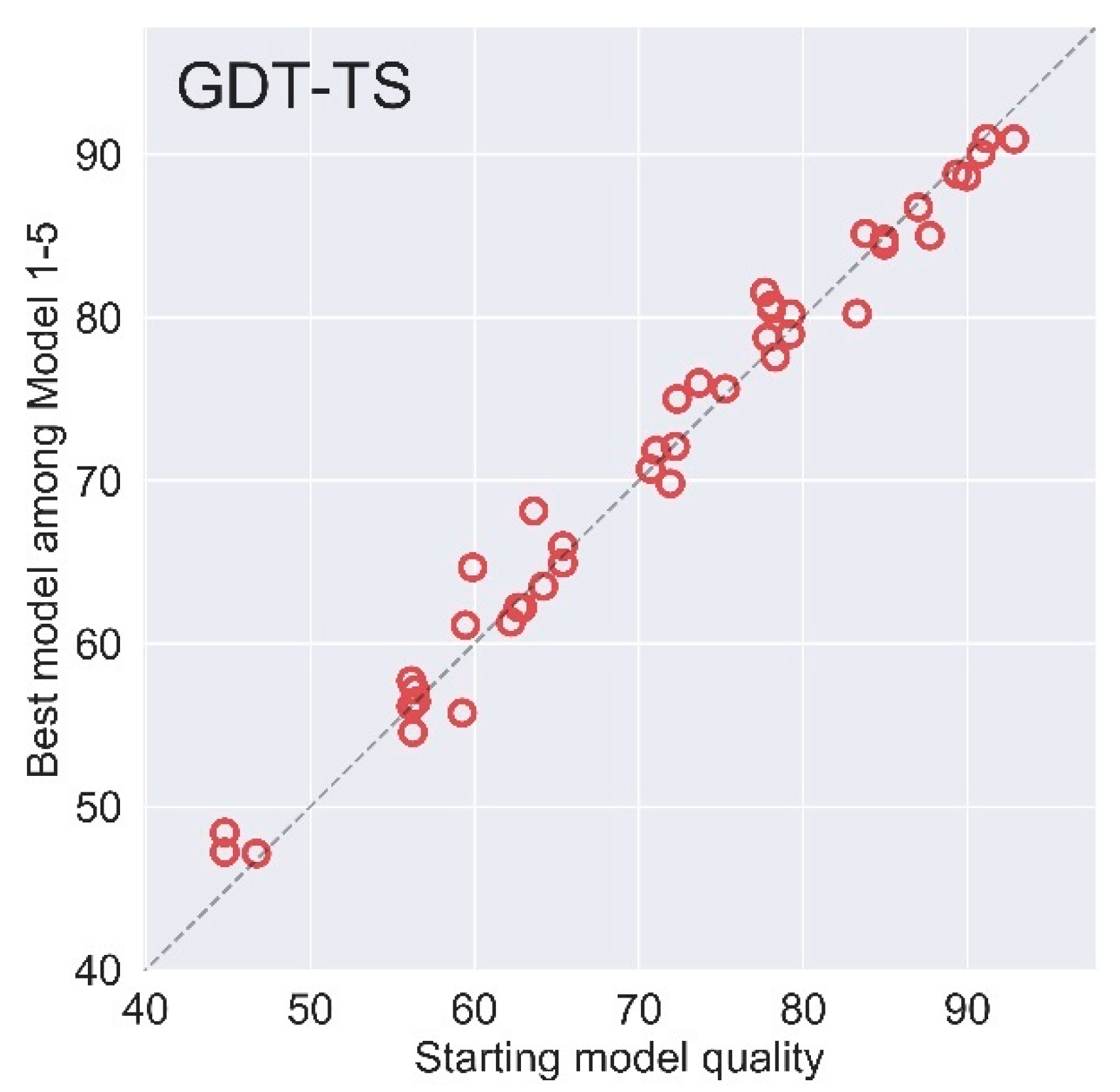
2.1. Effectiveness of AIR 2.0 on CASP13
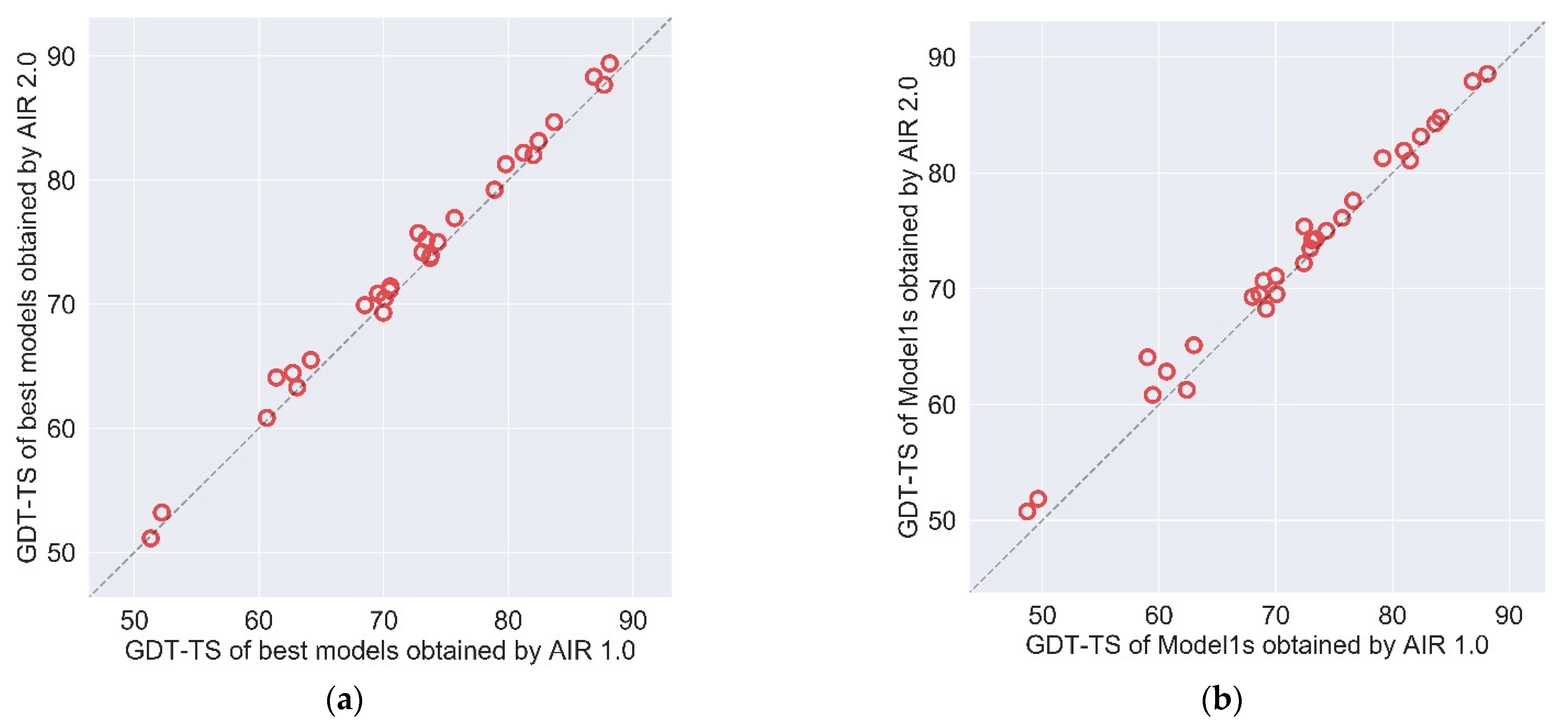
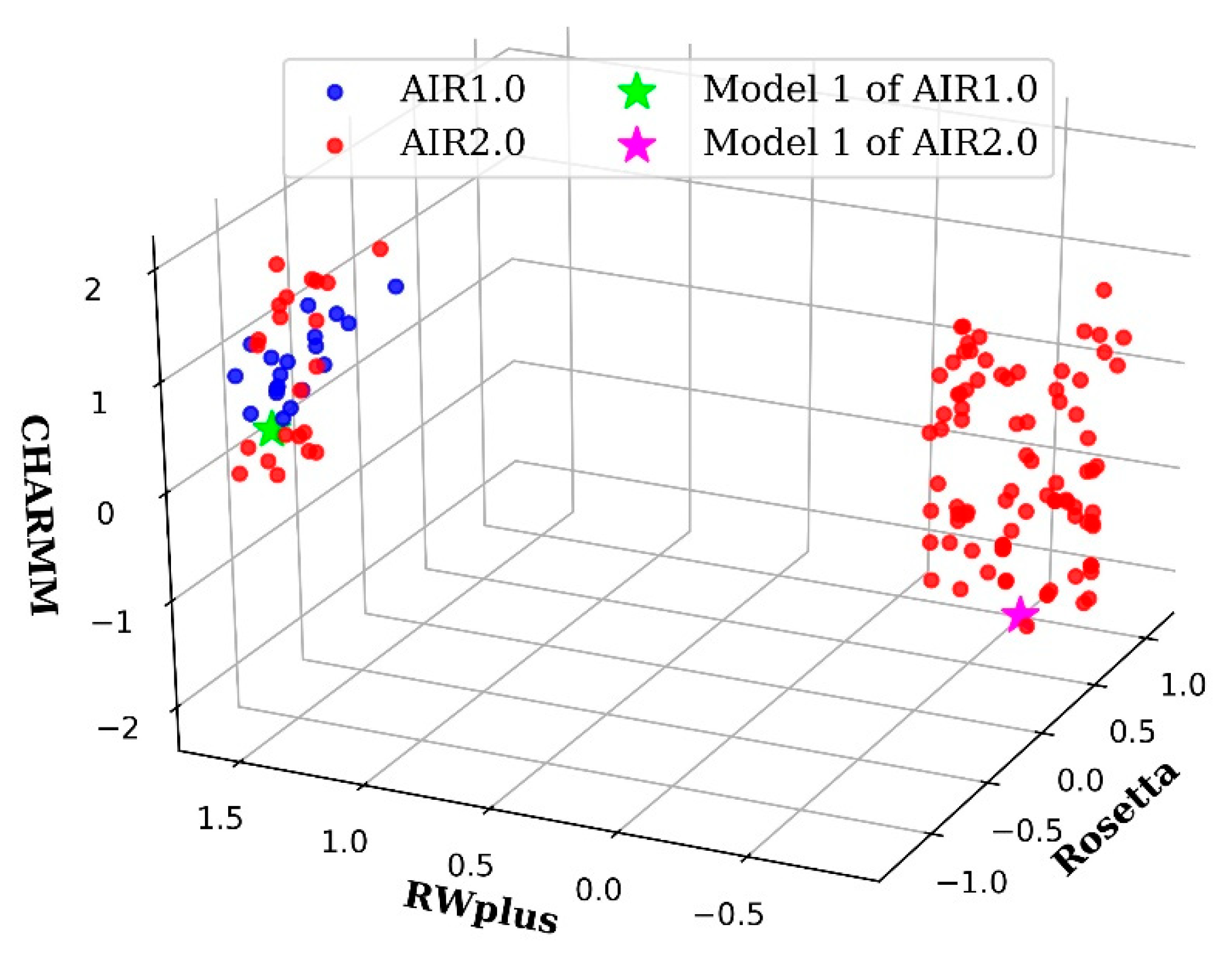
2.2. AIR 2.0 Is Superior to AIR 1.0
2.3. Comparison with Other State-of-the-Art Refinement Methods
2.4. Blind Test in CASP14
3. Discussion
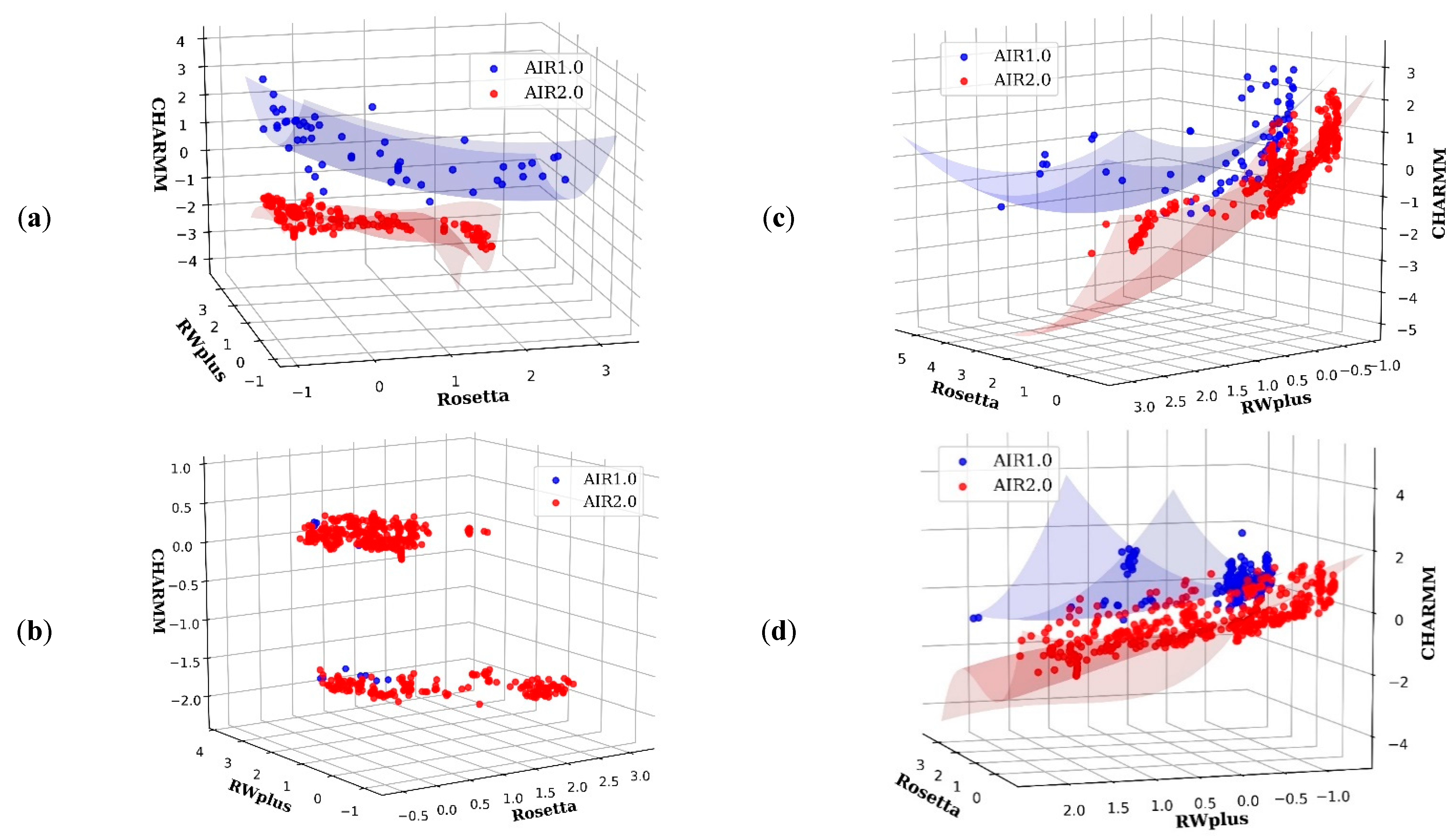
3.1. The Importance of the Diversity on AIR 2.0
3.2. The Influence of Hyperparameters on AIR 2.0
4. Methods
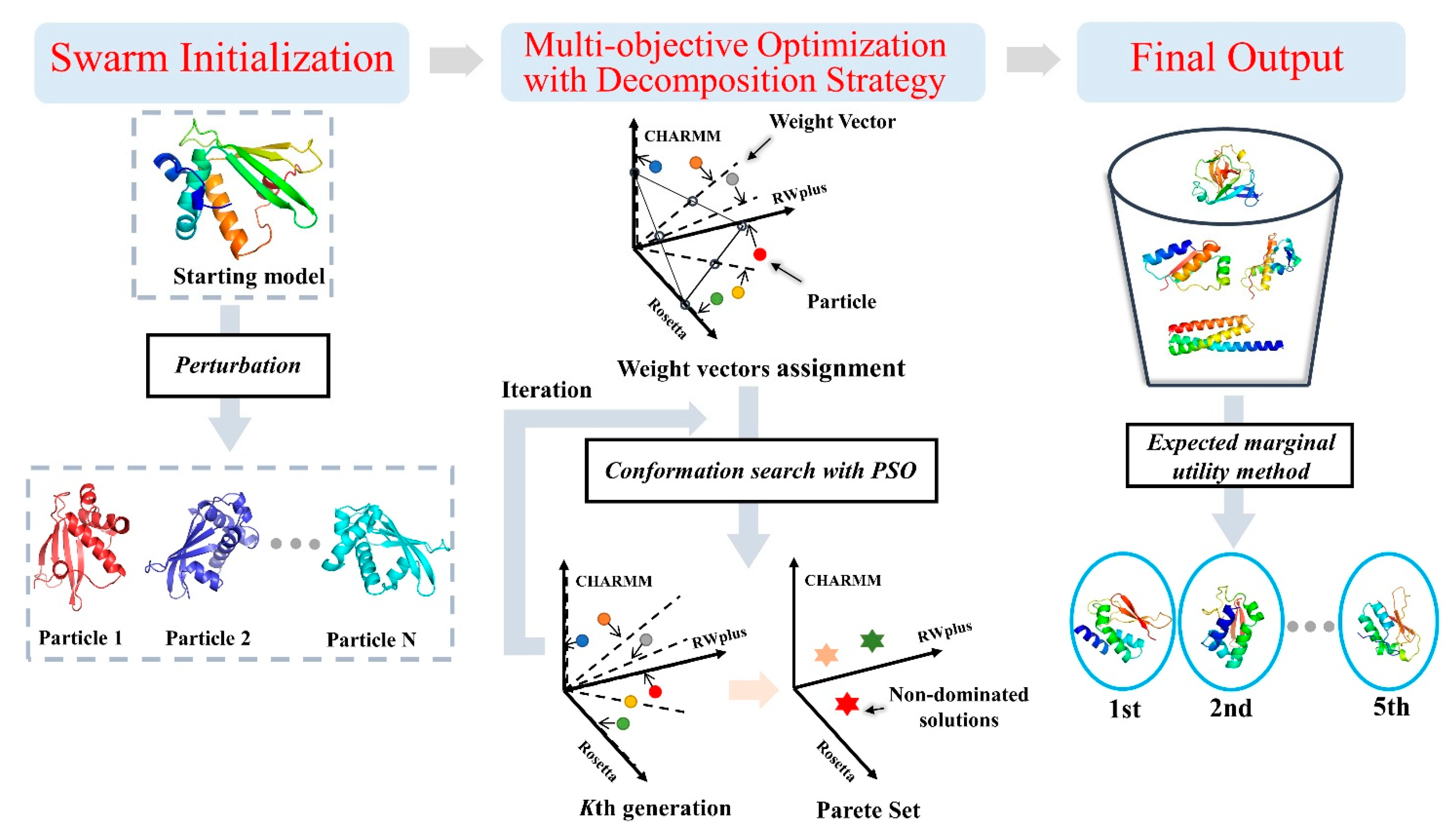
4.1. Overview of Refinement Pipeline AIR 2.0
4.2. Representations of Protein Conformations
4.3. Multi-Objective Optimization
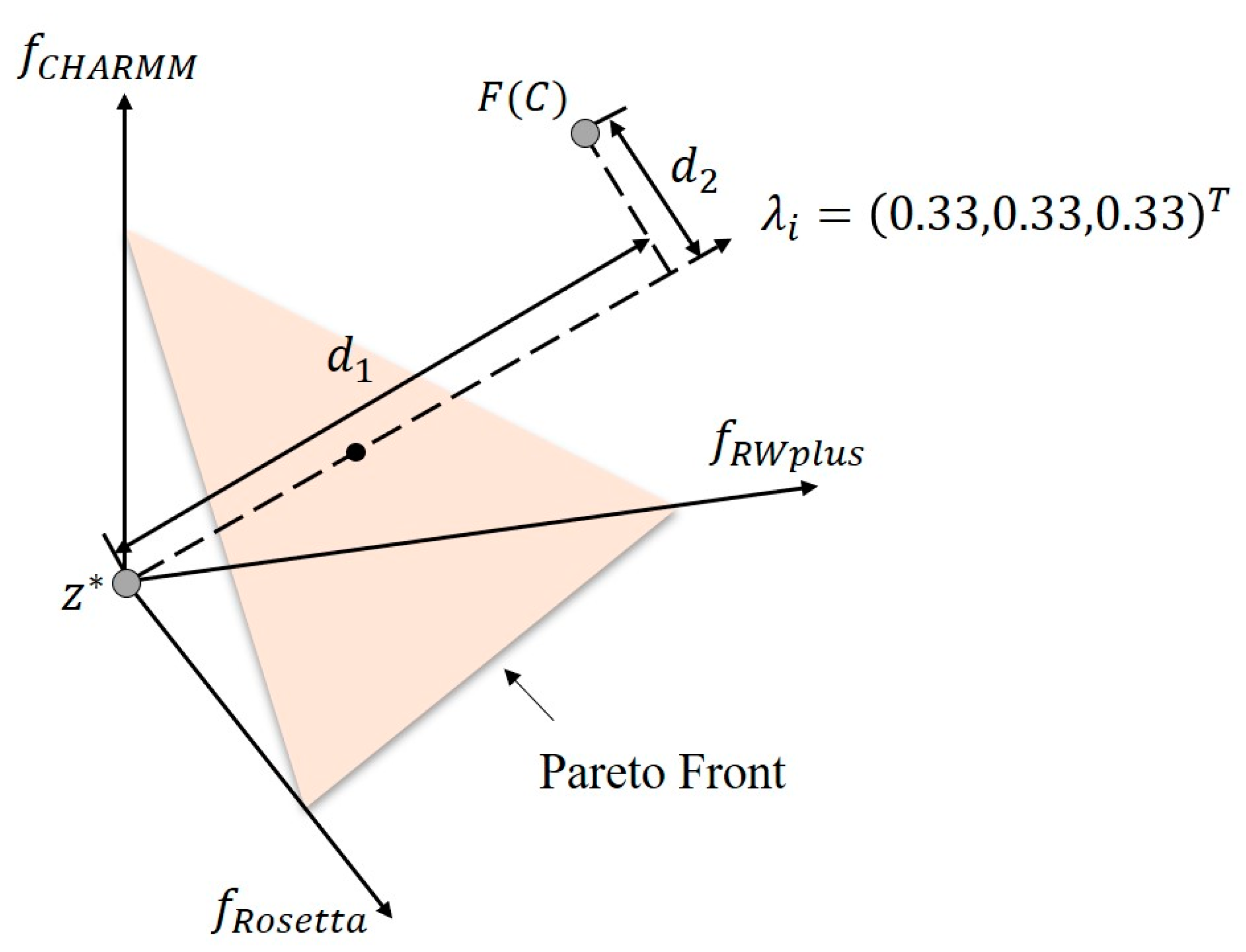
4.4. Decomposition Approach in Multi-Objective Optimization
4.5. Particle Swarm Optimization
4.6. Obtaining Pareto Optimal Set with Multi-Objective Particle Swarm Optimization Based on Decomposition Strategy
| Algorithm 1 Main Framework of AIR 2.0 |
| Input: Initial model , the maximum number of iterations MaxIT, the number of particles N. |
| Output: Pareto set . |
/*Initialization*/
|
4.7. Model Selection
5. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Availability and Implementation
References
- Schwede, T.; Peitsch, M.C. Computational Structural Biology: Methods and Applications; World Scientific: Singapore, 2008. [Google Scholar]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Kihara, D.; Lu, H.; Kolinski, A.; Skolnick, J. TOUCHSTONE: An ab initio protein structure prediction method that uses threading-based tertiary restraints. Proc. Natl. Acad. Sci. USA 2001, 98, 10125–10130. [Google Scholar] [CrossRef] [Green Version]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [Green Version]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renzhi, C.; Debswapna, B.; Badri, A.; Jilong, L.; Jianlin, C. Massive integration of diverse protein quality assessment methods to improve template based modeling in CASP11. Proteins Struct. Funct. Bioinform. 2015, 84, 247–259. [Google Scholar]
- Hou, J.; Wu, T.; Cao, R.; Cheng, J. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13. Proteins Struct. Funct. Bioinform. 2019, 87, 1165–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamisetty, H.; Ovchinnikov, S.; Baker, D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. USA 2013, 110, 15674–15679. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Guo, Z.; Hou, J.; Cheng, J. DeepDist: Real-value inter-residue distance prediction with deep residual convolutional network. BMC Bioinform. 2021, 22, 1–17. [Google Scholar] [CrossRef]
- Adhikari, B.; Cheng, J. CONFOLD2: Improved contact-driven ab initio protein structure modeling. BMC Bioinform. 2018, 19, 22. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, B.; Hou, J.; Cheng, J. Protein contact prediction by integrating deep multiple sequence alignments, coevolution and machine learning. Proteins Struct. Funct. Bioinform. 2018, 86, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Kandathil, S.M.; Greener, J.G.; Jones, D.T. Recent developments in deep learning applied to protein structure prediction. Proteins 2019, 87, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Won, J.; Heo, L.; Seok, C. GalaxyRefine2: Simultaneous refinement of inaccurate local regions and overall protein structure. Nucleic Acids Res. 2019, 47, W451–W455. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Adhikari, B.; Cheng, J. DeepSF: Deep convolutional neural network for mapping protein sequences to folds. Bioinformatics 2018, 34, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Arbour, C.F.; Feig, M. Driven to near-experimental accuracy by refinement via molecular dynamics simulations. Proteins 2019, 87, 1263–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovan, L.; Oleinikovas, V.; Yalinca, H.; Kryshtafovych, A.; Saladino, G.; Gervasio, F.L. Assessment of the model refinement category in CASP12. Proteins 2018, 86 (Suppl. 1), 152–167. [Google Scholar] [CrossRef] [Green Version]
- Modi, V.; Dunbrack, R.L., Jr. Assessment of refinement of template-based models in CASP11. Proteins 2016, 84 (Suppl. 1), 260–281. [Google Scholar] [CrossRef] [Green Version]
- Read, R.J.; Sammito, M.D.; Kryshtafovych, A.; Croll, T.I. Evaluation of model refinement in CASP13. Proteins 2019, 87, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Feig, M. Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2018, 115, 13276–13281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Lee, G.R.; Kim, D.E.; Anishchenko, I.; Cong, Q.; Baker, D. High-accuracy refinement using Rosetta in CASP13. Proteins 2019, 87, 1276–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terashi, G.; Kihara, D. Protein structure model refinement in CASP12 using short and long molecular dynamics simulations in implicit solvent. Proteins Struct. Funct. Bioinform. 2018, 86, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S. Development and testing of the OPLS all-atom force field on conformational energetics. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y. A Novel Side-Chain Orientation Dependent Potential Derived from Random-Walk Reference State for Protein Fold Selection and Structure Prediction. PLoS ONE 2010, 5, e15386. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11, 2714–2726. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Skolnick, J. GOAP: A Generalized Orientation-Dependent, All-Atom Statistical Potential for Protein Structure Prediction. Biophys. J. 2011, 101, 2043–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. PREFMD: A web server for protein structure refinement via molecular dynamics simulations. Bioinformatics 2018, 34, 1063–1065. [Google Scholar] [CrossRef] [Green Version]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein structure prediction using Rosetta. Methods Enzymol. 2003, 383, 66. [Google Scholar]
- Wang, D.; Geng, L.; Zhao, Y.J.; Yang, Y.; Huang, Y.; Zhang, Y.; Shen, H.B. Artificial intelligence-based multi-objective optimization protocol for protein structure refinement. Bioinformatics 2020, 36, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Chapman, R.; Dozier, G. ACM Press the 38th annual. In Proceedings of the 38th Annual on Southeast Regional Conference, ACM-SE 38, Multiobjective Particle Swarm Optimization, Clemson, SC, USA, 7–8 April 2000; p. 56. [Google Scholar]
- Deb, K.; Pratap, A.; Agarwal, S.; Meyarivan, T. A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Trans. Evol. Comput. 2002, 6, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Coello, C.A.C.; Pulido, G.T.; Lechuga, M.S. Handling multiple objectives with particle swarm optimization. IEEE Trans. Evol. Comput. 2004, 8, 256–279. [Google Scholar] [CrossRef]
- Hui, L.; Qingfu, Z. Multiobjective Optimization Problems with Complicated Pareto Sets, MOEA/D and NSGA-II. IEEE Trans. Evol. Comput. 2009, 13, 284–302. [Google Scholar] [CrossRef]
- Trivedi, A.; Srinivasan, D.; Sanyal, K.; Ghosh, A. A Survey of Multiobjective Evolutionary Algorithms based on Decomposition. IEEE Trans. Evol. Comput. 2016, 21, 440–462. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzetto, D.; Kryshtafovych, A.; Fidelis, K.; Moult, J.; Tramontano, A. Evaluation of template-based models in CASP8 with standard measures. Proteins Struct. Funct. Bioinform. 2009, 77 (Suppl. 9), 18–28. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Qingfu, Z.; Hui, L. MOEA/D: A Multiobjective Evolutionary Algorithm Based on Decomposition. IEEE Trans. Evol. Comput. 2007, 11, 712–731. [Google Scholar] [CrossRef]
- Cheng, R.; Jin, Y.; Olhofer, M.; Sendhoff, B. A Reference Vector Guided Evolutionary Algorithm for Many-Objective Optimization. IEEE Trans. Evol. Comput. 2016, 20, 773–791. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, A.; Omidvar, M.N.; Li, X.; Deb, K. Sensitivity analysis of Penalty-based Boundary Intersection on aggregation-based EMO algorithms. In Proceedings of the 2015 IEEE Congress on Evolutionary Computation (CEC), Sendai, Japan, 25–28 May 2015. [Google Scholar]
- Yang, S.; Jiang, S.; Jiang, Y. Improving the Multiobjective Evolutionary Algorithm Based on Decomposition with New Penalty Schemes. Soft Comput. 2016, 21, 4677–4691. [Google Scholar] [CrossRef] [Green Version]
- Das, I.; Dennis, J.E. Normal-Boundary Intersection: A New Method for Generating the Pareto Surface in Nonlinear Multicriteria Optimization Problems. Siam J. Opt. 1996, 8, 631–657. [Google Scholar] [CrossRef] [Green Version]
- Branke, J.; Deb, K.; Dierolf, H.; Osswald, M. Finding knees in multi-objective optimization. In International Conference on Parallel Problem Solving from Nature; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Parsons, J.; Holmes, J.B.; Rojas, J.M.; Tsai, J.; Strauss, C.E. Practical conversion from torsion space to Cartesian space for in silico protein synthesis. J. Comput. Chem. 2005, 26, 1063–1068. [Google Scholar] [CrossRef]
- AlQuraishi, M. Parallelized Natural Extension Reference Frame: Parallelized Conversion from Internal to Cartesian Coordinates. J. Comput. Chem. 2019, 40, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kavraki, L.E. A New Method for Fast and Accurate Derivation of Molecular Conformations. J. Chem. Inform. Model. 2002, 42, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.K.; Bandyopadhyay, S.; Pal, S.K. Multi-Objective Particle Swarm Optimization with time variant inertia and acceleration coefficients. Inform. Sci. 2007, 177, 5033–5049. [Google Scholar] [CrossRef] [Green Version]
- Zapotecas Martínez, S.; Coello Coello, C.A. A multi-objective particle swarm optimizer based on decomposition. In Proceedings of the 13th Annual Conference on Genetic and Evolutionary Computation, Dublin, Ireland, 12–16 July 2011; p. 69. [Google Scholar]
- Miettinen, K. Nonlinear Multiobjective Optimization; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Kennedy, J.; Eberhart, R. Particle Swarm Optimization. In Book Particle Swarm Optimization; BoD—Books on Demand GmbH: Norderstedt, Germany, 2002. [Google Scholar]
- Parsopoulos, K.E.; Vrahatis, M.N. Particle swarm optimization method in multiobjective problems. In Proceedings of the 2002 ACM Symposium on Applied Computing, Madrid, Spain, 10–14 March 2002. [Google Scholar]
- Wallner, B.; Fang, H.; Elofsson, A. Automatic consensus-based fold recognition using Pcons, ProQ, and Pmodeller. Proteins Struct. Funct. Bioinform. 2003, 53, 534–541. [Google Scholar] [CrossRef]
- Kim, H.; Kihara, D. Detecting local residue environment similarity for recognizing near-native structure models. Proteins 2014, 82, 3255–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat. Commun. 2021, 12, 1340. [Google Scholar] [CrossRef] [PubMed]
- Uziela, K.; Menendez Hurtado, D.; Shu, N.; Wallner, B.; Elofsson, A. ProQ3D: Improved model quality assessments using deep learning. Bioinformatics 2017, 33, 1578–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Best Model (GDT-TS) | Model 1 (GDT-TS) |
|---|---|---|
| AIR 1.0 | 1.07 | 0.16 |
| AIR 2.0 | 1.98 | 1.22 |
| Target | Initial Model | AIR 2.0 | BAKER | FEIGLAB | Zhang |
|---|---|---|---|---|---|
| R0949 | 64.53 | 65.12 | 56.01 | 62.98 | 64.53 |
| R0957 | 60.97 | 64.08 | 60.32 | 61.45 | 61.61 |
| R0968s1 | 66.74 | 69.71 | 78.81 | 72.25 | 69.07 |
| R0974s1 | 84.78 | 85.96 | 99.64 | 97.10 | 84.06 |
| R0976D2 | 83.06 | 84.27 | 89.11 | 80.64 | 83.87 |
| R0979 | 70.65 | 74.18 | 60.60 | 70.38 | 70.38 |
| R0986s1 | 80.16 | 83.15 | 90.76 | 93.21 | 77.99 |
| R0989D1 | 50.75 | 51.12 | 44.22 | 50.75 | N/A |
| R0999D3 | 75.14 | 76.94 | 76.11 | 76.94 | 74.31 |
| R1002D2 | 88.14 | 88.56 | 89.41 | 79.24 | 88.14 |
| R1004D2 | 78.57 | 77.60 | 81.49 | 93.51 | 79.22 |
| R1016 | 81.06 | 82.11 | 78.22 | 81.68 | 80.45 |
| Target | Length | ||||
|---|---|---|---|---|---|
| R0974s1 | 69 | 0.27 | 1.18 | 0.81 | 1.24 |
| R1004D2 | 77 | 1.05 | 0.97 | 0.98 | |
| R0968s1 | 118 | 2.58 | 2.97 | 3.18 | 2.94 |
| R0981D5 | 127 | 0.79 | 0.59 | 0.44 | 0.20 |
| R0959 | 189 | 3.30 | 3.83 | 3.97 | 3.74 |
| R0981D3 | 203 | 0.49 | 0.13 | 0 | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, C.-P.; Wang, D.; Pan, X.; Shen, H.-B. Protein Structure Refinement Using Multi-Objective Particle Swarm Optimization with Decomposition Strategy. Int. J. Mol. Sci. 2021, 22, 4408. https://doi.org/10.3390/ijms22094408
Zhou C-P, Wang D, Pan X, Shen H-B. Protein Structure Refinement Using Multi-Objective Particle Swarm Optimization with Decomposition Strategy. International Journal of Molecular Sciences. 2021; 22(9):4408. https://doi.org/10.3390/ijms22094408
Chicago/Turabian StyleZhou, Cheng-Peng, Di Wang, Xiaoyong Pan, and Hong-Bin Shen. 2021. "Protein Structure Refinement Using Multi-Objective Particle Swarm Optimization with Decomposition Strategy" International Journal of Molecular Sciences 22, no. 9: 4408. https://doi.org/10.3390/ijms22094408
APA StyleZhou, C.-P., Wang, D., Pan, X., & Shen, H.-B. (2021). Protein Structure Refinement Using Multi-Objective Particle Swarm Optimization with Decomposition Strategy. International Journal of Molecular Sciences, 22(9), 4408. https://doi.org/10.3390/ijms22094408