
Mechanisms of High-Grade Serous Carcinogenesis in the Fallopian Tube and Ovary: Current Hypotheses, Etiologic Factors, and Molecular Alterations


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
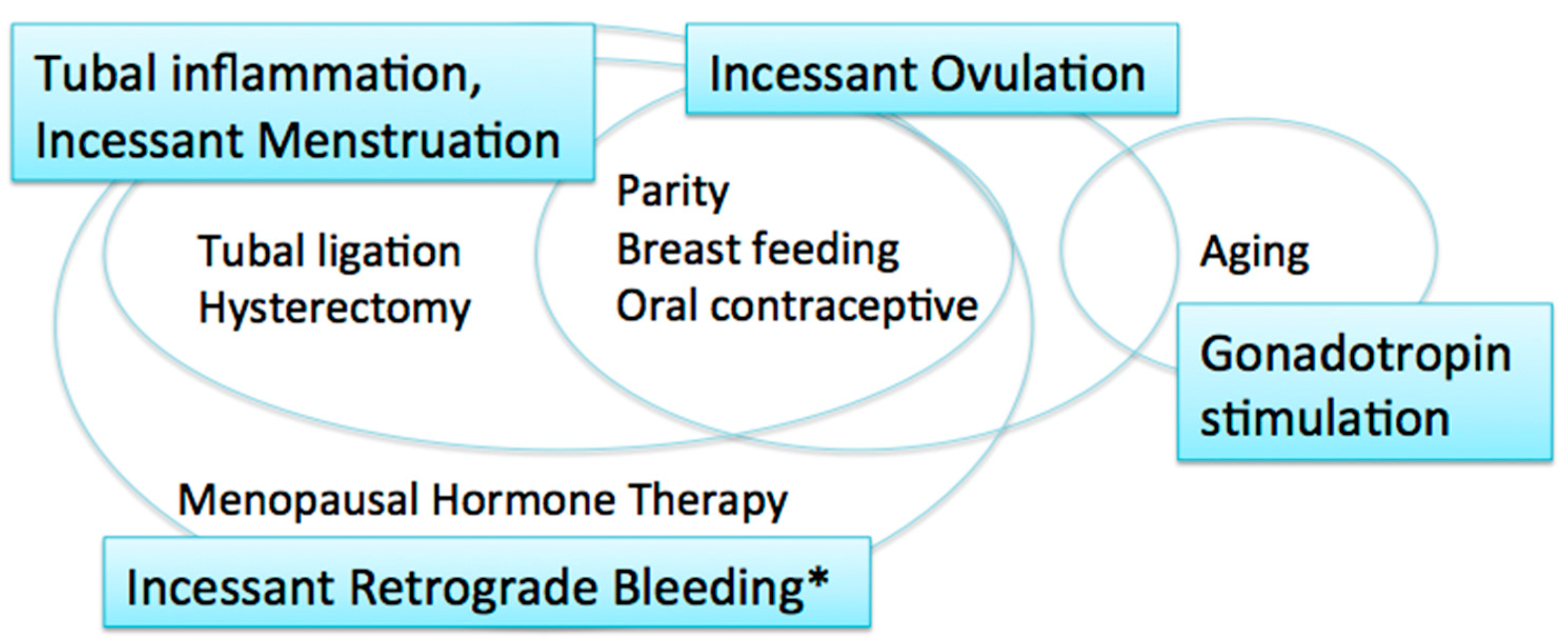
2. Carcinogenic Hypotheses and Risk/Protective Factors
2.1. Incessant Ovulation
2.2. Gonadotropin Stimulation
2.3. Tubal Inflammation
2.4. Incessant Menstruation
2.5. Incessant Retrograde Bleeding
3. Sites of Origin of HGSCs
3.1. Fallopian Tube Epithelium
3.2. Ovarian Surface Epithelium
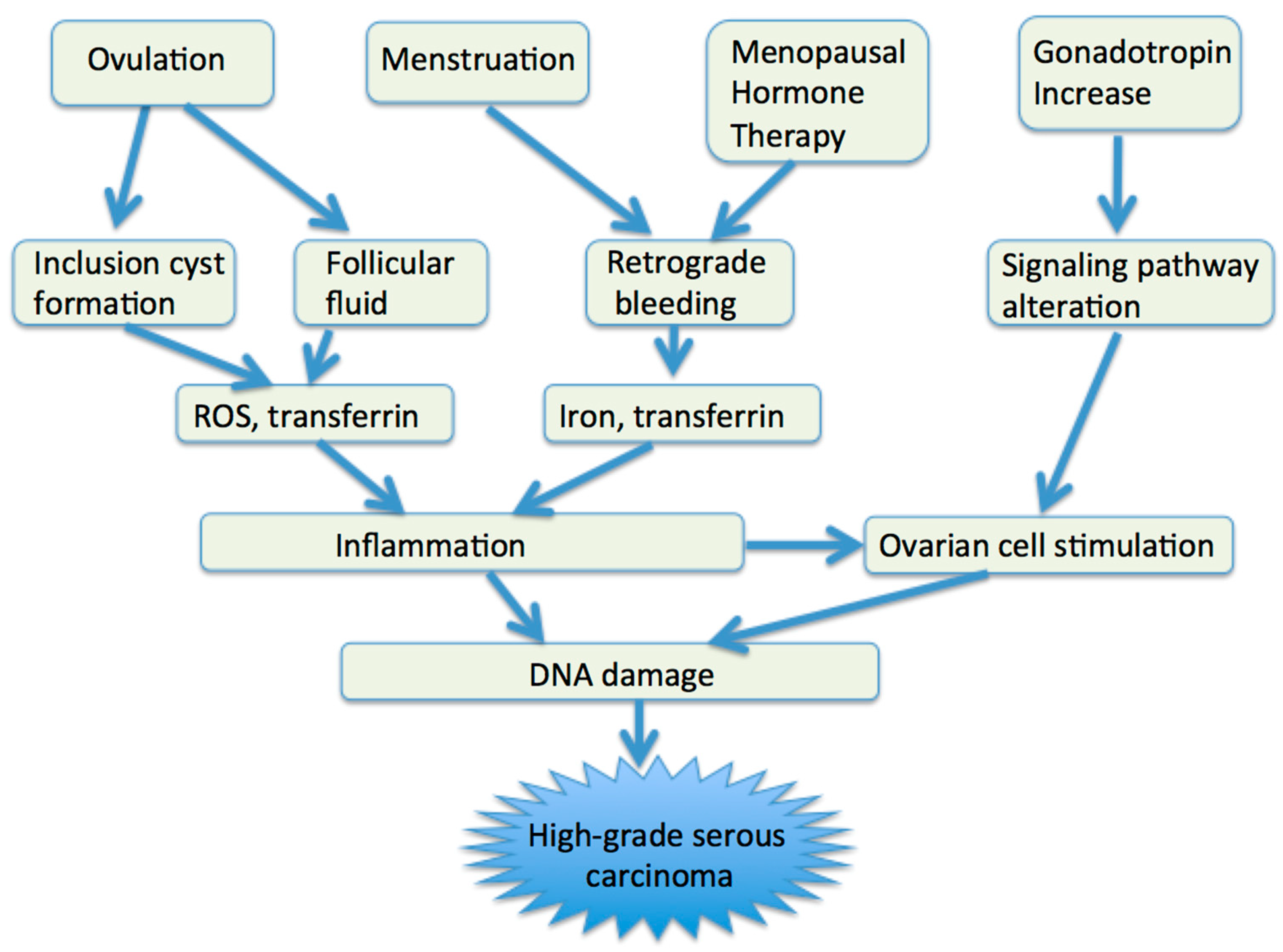
4. Etiologic Factors
4.1. Ovulation
4.2. Retrograde Bleeding
4.3. Gonadotropin
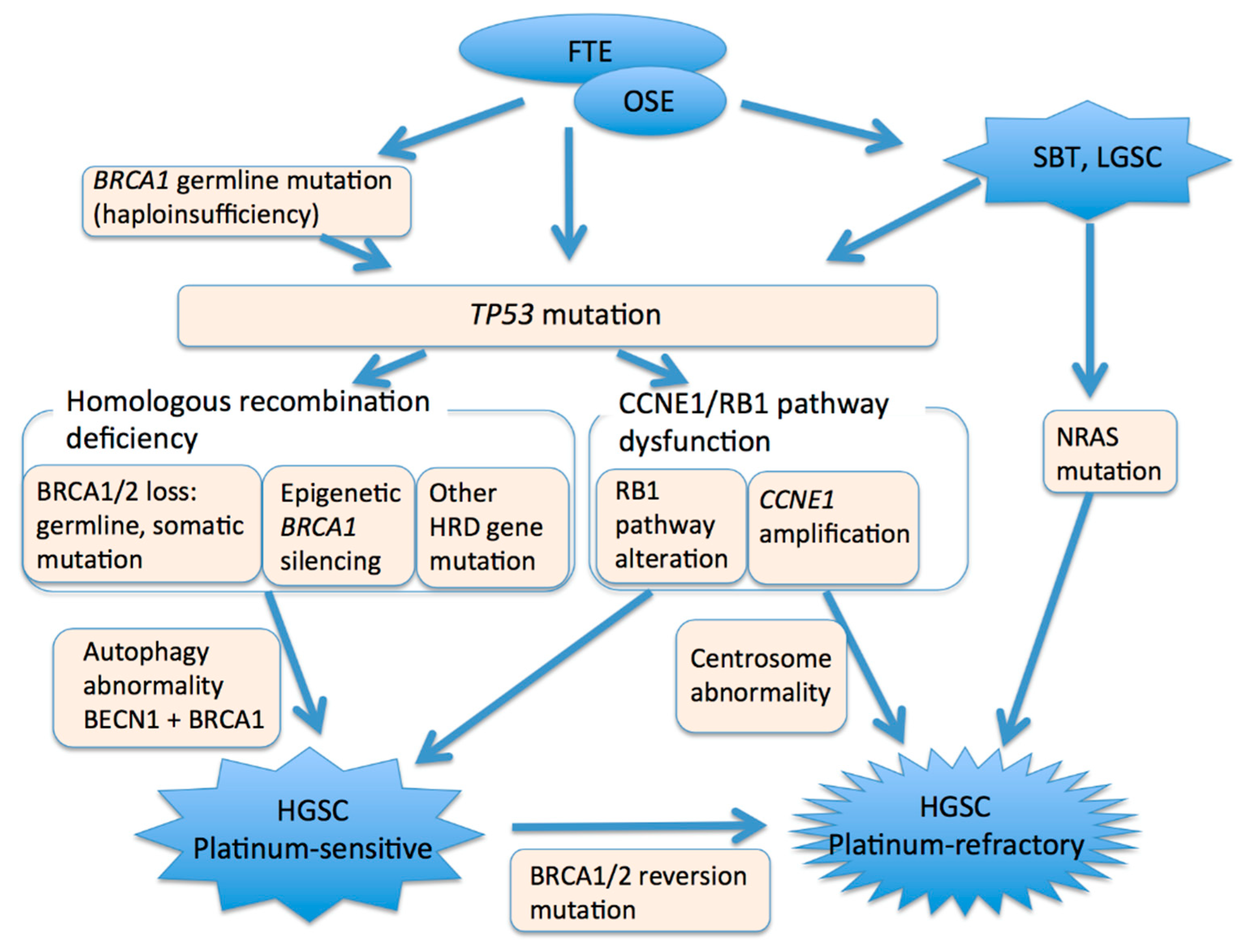
5. Molecular Alterations
5.1. TP53
5.2. BRCA1/2
5.3. CCNE1 and Rb1
5.4. Autophagy Gene
5.5. Stem Cell Markers
5.6. Transition from Low-Grade Serous Carcinoma
6. Clinical Relevance
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian carcinomas: At least five different diseases with distinct histological features and molecular genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Salazar, C.; Campbell, I.G.; Gorringe, K.L. When Is “Type I” Ovarian Cancer Not “Type I”? Indications of an Out-Dated Dichotomy. Front. Oncol. 2018, 8, 654. [Google Scholar] [CrossRef]
- Bowtell, D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef]
- Bodurka, D.C.; Deavers, M.T.; Tian, C.; Sun, C.C.; Malpica, A.; Coleman, R.L.; Lu, K.H.; Sood, A.K.; Birrel, M.J.; Ozols, R.; et al. Reclassification of serous ovarian carcinoma by a 2-tier system. A Gynecologic Oncology Group study. Cancer 2012, 119, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Woo, M.M.; Gilks, C.B. The origin of ovarian carcinomas: A developmental view. Gynecol. Oncol. 2008, 110, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Salvador, S.; Gilks, B.; Köbel, M.; Huntsman, D.; Rosen, B.; Miller, D. The fallopian tube: Primary site of most pelvic high-grade serous carcinomas. Int. J. Gynecol. Cancer 2009, 19, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Karpinskyj, C.; Gentry-Maharaj, A. Ovarian Cancer Prevention and Screening. Obstet Gynecol. 2018, 131, 909–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathalla, M.F. Incessant ovulation—A factor in ovarian neoplasia? Lancet 1971, 2, 163. [Google Scholar] [CrossRef]
- Schildkraut, J.M.; Bastos, E.; Berchuck, A. Relationship between lifetime ovulatory cycles and overexpression of mutant p53 in epithelial ovarian cancer. J. Natl. Cancer Inst. 1997, 89, 932–938. [Google Scholar] [CrossRef] [Green Version]
- Stadel, B.V. The etiology and prevention of ovarian cancer. Am. J. Obstet Gynecol. 1975, 123, 772–774. [Google Scholar] [CrossRef]
- Mertens-Walker, I.; Baxter, R.C.; Marsh, D.J. Gonadotropin signalling in epithelial ovarian cancer. Cancer Lett. 2012, 324, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Siristatidis, C.T.N.; Sergentanis, P.; Kanavidis, P.; Trivella, M.; Sotiraki, M.; Marvromatis, I.; Psaltopoulou, T.; Skalkidou, A.; Petridou, E.T. Controlled ovarian hyperstimulation for IVF: Impact on ovarian, endometrial and cervical cancer—A systematic review and meta-analysis. Hum. Reprod Update 2013, 19, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Gronwald, J.; Glass, K.; Rosen, B.; Karlan, B.; Tung, N.; Neuhausen, S.L.; Moller, P.; Ainsworth, P.; Sun, P.; Narod, S.A.; et al. Treatment of infertility does not increase the risk of ovarian cancer among women with a BRCA1 or BRCA2 mutation. Fertil Steril 2016, 105, 781–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.R.; Xu, X.X. Etiology of epithelial ovarian cancer: A cellular mechanism for the role of gonadotropins. Gynecol. Oncol. 2003, 91, 1–2. [Google Scholar] [CrossRef]
- Vercellini, P.; Crosignani, P.; Somigliana, E.; Vigano, P.; Buggio, L.; Bolis, G.; Fedele, L. The incessant menstruation’ hypothesis: A mechanistic ovarian cancer model with implications for prevention. Hum. Reprod 2011, 26, 2262–2273. [Google Scholar] [CrossRef] [Green Version]
- Crum, C.P.; Drapkin, R.; Kindelberger, D.; Medeiros, F.; Miran, A.; Lee, Y. Lessons from BRCA: The tubal fimbria emerges as an origin for pelvic serous cancer. Clin. Med. Res. 2007, 5, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Idahl, A.; Le Cornet, C.; Maldonado, S.G.; Waterboer, T.; Bender, N.; Tjønneland, A.; Hansen, L.; Boutron-Ruault, M.-C.; Fournier, A.; Kvaskoff, M.; et al. Serologic markers of Chlamydia trachomatis and other sexually transmitted infections and subsequent ovarian cancer risk: Results from the EPIC cohort. Int. J. Cancer 2020, 147, 2042–2052. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, I.; Matsuura, T. Screening and prevention for high-grade serous carcinoma of the ovary based on carcinogenesis—Fallopian tube- and ovarian-derived tumors and incessant retrograde bleeding. Diagnostics 2020, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Riman, T.; Dickman, P.W.; Nilsson, S.; Correia, N.; Nordlinder, H.; Magnusson, C.M.; Weiderpass, E.; Persson, I.R. Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J. Natl Cancer Inst. 2002, 94, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Koskela-Niska, V.; Riska, A.; Lyytinen, H.; Pukkala, E.; Ylikorkala, O. Primary fallopian tube carcinoma risk in users of postmenopausal hormone therapy in Finland. Gynecol. Oncol. 2012, 126, 241–244. [Google Scholar] [CrossRef]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Shaw, P.A.; Kurman, R.J.; Shih, I.M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Tan, S.; Singh, G.; Rizk, P.; Swathi, Y.; Tan, T.Z.; Huang, R.Y.; Leushacke, M.; Barker, N. Lgr5 marks stem/progenitor cells in ovary and tubal epithelia. Nat. Cell. Biol. 2014, 16, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Hao, D.; Li, J.; Jia, S.; Meng, Y.; Zhang, C.; Di, L.-J. Integrated analysis reveals tubal- and ovarian-originated serous ovarian cancer and predicts differential therapeutic responses. Clin. Cancer Res. 2017, 23, 7400–7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Dolgalev, I.; Zhang, T.; Ran, H.; Levine, D.A.; Neel, B.G. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 2019, 10, 5367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geistlinger, L.; Oh, S.; Ramos, M.; Schiffer, L.; Larue, R.S.; Henzler, C.M.; Munro, S.A.; Daughters, C.; Nelson, A.C.; Winterhoff, B.J.; et al. Multiomic Analysis of Subtype Evolution and Heterogeneity in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020, 80, 4335–4345. [Google Scholar] [CrossRef]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef] [Green Version]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.L.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 2012, 72, 4141–4153. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, D.Y.; Janzen, D.M.; Schafenacker, A.M.; Velasco, V.S.; Shung, M.S.; Chieng, D.; Huang, J.; Witte, O.N.; Memarzadeh, S. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: A site for injury and serous cancer initiation. Stem Cells 2012, 30, 2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flesken-Nikitin, A.; Hwang, C.I.; Cheng, C.Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Crum, C.P. Intercepting pelvic cancer in the distal fallopian tube: Theories and realities. Mol. Oncol. 2009, 3, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Meyer, L.A.; Deavers, M.T.; Daniels, M.S.; Keeler, E.R.; Mok, S.C.; Gershenson, D.M.; Lu, K.H. Microscopic and early-stage ovarian cancers in BRCA1/2 mutation carriers: Building a model for early BRCA-associated tumorigenesis. Cancer Prev. Res. 2011, 4, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilks, C.B.; Irving, J.; Köbel, M.; Lee, C.; Singh, N.; Wilkinson, N.; McCluggage, W.G. Incidental Nonuterine High-grade Serous Carcinomas Arise in the Fallopian Tube in Most Cases Further Evidence for the Tubal Origin of High-grade Serous Carcinomas. Am. J. Surg Pathol. 2015, 39, 357–364. [Google Scholar] [CrossRef]
- Xiang, L.; Rong, G.; Zhao, J.; Wang, Z.; Shi, F. Identification of candidate genes associated with tubal origin of high-grade serous ovarian cancer. Oncol. Lett. 2018, 15, 7769–7775. [Google Scholar] [CrossRef] [Green Version]
- Beirne, J.P.; McArt, D.G.; Roddy, A.; McDermott, C.; Ferris, J.; Buckley, N.E.; Coulter, P.; McCabe, N.; Eddie, S.I.; McCluggage, W.G.; et al. Defining the molecular evolution of extrauterine high grade serous carcinoma. Gynecol. Oncol. 2019, 155, 305–317. [Google Scholar] [CrossRef]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef]
- Xian, W.; Miron, A.; Roh, M.; Semmel, D.R.; Yassin, Y.; Garber, J.; Oliva, E.; Goodman, A.; Mehra, K.; Berkowitz, R.S.; et al. The Li-Fraumeni syndrome (LFS): A model for the initiation of p53 signatures in the distal Fallopian tube. J. Pathol. 2010, 220, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Asaka, S.; Davis, C.; Lin, S.-F.; Wang, T.-L.; Heaphy, C.M.; Shih, I.-M. Analysis of Telomere Lengths in p53 Signatures and Incidental Serous Tubal Intraepithelial Carcinomas without Concurrent Ovarian Cancer. Am. J. Surg. Pathol. 2019, 43, 1083–1091. [Google Scholar] [CrossRef]
- Mehra, K.K.; Chang, M.C.; Folkins, A.K.; Raho, C.J.; Lima, J.F.; Yuan, L.; Mehrad, M.; Tworoger, S.S.; Crum, C.P.; Saleemuddin, A. The impact of tissue block sampling on the detection of p53 signatures in fallopian tubes from women with BRCA 1 or 2 mutations (BRCA+) and controls. Mod. Pathol. 2011, 24, 152–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, L.C.; Kafkova, S.; Leonhardt, K.; Kellner, C.; Einenkel, J. Serous tubal in situ carcinoma (STIC) in primary peritoneal serous carcinomas. Int. J. Gynecol. Pathol. 2013, 32, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Gaitskell, K.; Garcia, M.J.; Albukhari, A.; Tsaltas, J.; Ahmed, A.A. Serous tubal intraepithelial carcinomas associated with high-grade serous ovarian carcinomas: A systematic review. BJOG 2017, 124, 872–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of ovarian cancer progression reveals diverse metastatic trajectories including intraepithelial metastasis to the fallopian tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Meserve, E.E.; Strickland, K.C.; Miron, A.; Soong, T.R.; Campbell, F.; Howitt, B.E.; Crum, C.P. Evidence of a Monoclonal Origin for Bilateral Serous Tubal Intraepithelial Neoplasia. Int. J. Gynecol. Pathol. 2019, 38, 443–448. [Google Scholar] [CrossRef]
- Visvanathan, K.; Shaw, P.; May, B.J.; Bahadirli-Talbott, A.; Kaushiva, A.; Risch, H.; Narod, S.; Wang, T.L.; Parkash, V.; Vang, R.; et al. Fallopian Tube Lesions in Women at High Risk for Ovarian Cancer: A. Multicenter Study. Cancer Prev. Res. 2018, 11, 697–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werness, B.A.; Parvatiyar, P.; Ramus, S.J.; Whittemore, A.S.; Garlinghouse-Jones, K.; Oakley-Girvan, I.; DiCioccio, R.A.; Wiest, J.; Tsukada, Y.; Ponder, B.A.J.; et al. Ovarian carcinoma in situ with germline BRCA1 mutation and loss of heterozygosity at BRCA1 and TP53. J. Natl. Cancer Inst. 2000, 92, 1088–1091. [Google Scholar] [CrossRef] [Green Version]
- Pothuri, B.; Leitao, M.M.; Levine, D.A.; Viale, A.; Olshen, A.B.; Arroyo, O.; Bogomolniy, F.; Olvera, N.; Lin, O.; Soslow, R.A.; et al. Genetic analysis of the early natural history of epithelial ovarian carcinoma. PLoS ONE 2010, 5, e10358. [Google Scholar] [CrossRef] [PubMed]
- Soong, T.R.; Howitt, B.E.; Miron, A.; Horowitz, N.S.; Campbell, F.; Feltmate, C.M.; Muto, M.G.; Berkowitz, R.S.; Nucci, M.R.; Xian, W.; et al. Evidence for lineage continuity between early serous proliferations (ESPs) in the Fallopian tube and disseminated high-grade serous carcinomas. J. Pathol. 2018, 246, 344–351. [Google Scholar] [CrossRef]
- Banet, N.; Kurman, R.J. Two Types of Ovarian Cortical Inclusion Cysts: Proposed Origin and Possible Role in Ovarian Serous Carcinogenesis. Int. J. Gynecol. Pathol. 2015, 34, 3–8. [Google Scholar] [CrossRef]
- Park, K.J.; Patel, P.; Linkov, I.; Jotwani, A.; Kauff, N.; Pike, M.C. Observations on the origin of ovarian cortical inclusion cysts in women undergoing risk-reducing salpingo-oophorectomy. Histopathology 2018, 72, 766–776. [Google Scholar] [CrossRef]
- Wang, Y.; Sessine, M.S.; Zhai, Y.; Tipton, C.; McCool, K.; Kuick, R.; Connolly, D.C.; Fearon, E.R.; Cho, K.R. Lineage tracing suggests that ovarian endosalpingiosis does not result from escape of oviductal epithelium. J. Pathol. 2019, 249, 206–214. [Google Scholar] [CrossRef]
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef]
- Quinn, B.A.; Brake, T.; Hua, X.; Baxter-Jones, K.; Litwin, S.; Ellenson, L.H.; Connolly, D.C. Induction of ovarian leiomyosarcomas in mice by conditional inactivation of Brca1 and p53. PLoS ONE 2009, 4, e8404. [Google Scholar] [CrossRef] [Green Version]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backman, S.; Kollara, A.; Haw, R.; Stein, L.; Brown, T.J. Glucocorticoid-induced reversal of interleukin-1β-stimulated inflammatory gene expression in human oviductal cells. PLoS ONE 2014, 9, e97997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.S.; Chu, S.C.; Hsu, C.F.; Chen, P.-C.; Ding, D.-C.; Chang, M.-Y.; Chu, T.-Y. Mutagenic, surviving and tumorigenic effects of follicular fluid in the context of p53 loss: Initiation of fimbria carcinogenesis. Carcinogenesis 2015, 36, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.F.; Gerry, E.; Shih, I.-M. Tubal origin of ovarian cancer–the double-edged sword of haemoglobin. J. Pathol. 2017, 242, 3–6. [Google Scholar] [CrossRef]
- Brand, H.; Barnabas, G.D.; Sapoznik, S.; Bahar-Shany, K.; Pozniak, Y.; Yung, Y.; Hourvitz, A.; Geiger, T.; Jacob-Hirsch, J.; Levanon, K. NF-κB-miR-155 axis activation mediates ovulation-induced oncogenic effects in fallopian tube epithelium. Carcinogenesis 2020, 2, bgaa068. [Google Scholar]
- Hsu, C.F.; Huang, H.S.; Chen, P.C.; Ding, D.C.; Chu, T.Y. IGF-axis confers transformation and regeneration of fallopian tube fimbria epithelium upon ovulation. EBioMedicine 2019, 41, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savant, S.S.; Sriramkumar, S.; O’Hagan, H.M. The Role of Inflammation and Inflammatory Mediators in the Development, Progression, Metastasis, and Chemoresistance of Epithelial Ovarian Cancer. Cancers 2018, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.Y.; Huang, H.S.; Chao, T.H.; Chou, H.M.; Fang, C.; Qin, C.Z.; Lin, C.Y.; Chu, T.Y.; Zhou, H.H. Progesterone Prevents High-Grade Serous Ovarian Cancer by Inducing Necroptosis of p53-Defective Fallopian Tube Epithelial Cells. Cell Rep. 2017, 18, 2557–2565. [Google Scholar] [CrossRef]
- DastranjTabrizi, A.; MostafaGharabaghi, P.; SheikhzadehHesari, F.; Sadeghi, L.; Zamanvandi, S.; Sarbakhsh, P.; Ghojazadeh, M. Impact and mechanistic role of oral contraceptive pills on the number and epithelial type of ovarian cortical inclusion cysts; a clinicopathology and immunohistochemical study. Diagn Pathol. 2016, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeta, S.; Toyoshima, M.; Kitatani, K.; Ishibashi, M.; Usui, T.; Yaegashi, N. Transferrin facilitates the formation of DNA double-strand breaks via transferrin receptor 1: The possible involvement of transferrin in carcinogenesis of high-grade serous ovarian cancer. Oncogene 2016, 35, 3577–3586. [Google Scholar] [CrossRef]
- Huang, H.S.; Hsu, C.F.; Chu, S.C.; Chen, P.C.; Ding, D.C.; Chang, M.Y.; Chu, T.Y. Haemoglobin in pelvic fluid rescues Fallopian tube epithelial cells from reactive oxygen species stress and apoptosis. J. Pathol. 2016, 240, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Soria-Valles, C.; López-Soto, A.; Osorio, F.G.; López-Otín, C. Immune and inflammatory responses to DNA damage in cancer and aging. Mech. Ageing Dev. 2017, 165, 10–16. [Google Scholar] [CrossRef]
- Zheng, W.; Lu, J.J.; Luom, F.; Zheng, Y.; Feng, Y.-J.; Felix, J.C.; Lauchlan, S.C.; Pike, M.C. Ovarian Epithelial Tumor Growth Promotion by Follicle-Stimulating Hormone and Inhibition of the Effect by Luteinizing Hormone. Gynecol. Oncol. 2000, 76, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jin, H.; Liu, Y.; Zhou, J.; Ding, J.; Cheng, K.W.; Yu, Y.; Feng, Y. FSH inhibits ovarian cancer cell apoptosis by up-regulating survivin and down-regulating PDCD6 and DR5. Endocr Relat. Cancer 2010, 18, 13–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zheng, T.; Hong, W.; Ye, H.; Hu, C.; Zheng, Y. Mechanism for the Decision of Ovarian Surface Epithelial Stem Cells to Undergo Neo-Oogenesis or Ovarian Tumorigenesis. Cell Physiol. Biochem. 2018, 50, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Xu, X.X. Ovarian ageing, follicle depletion, and cancer: A hypothesis for the aetiology of epithelial ovarian cancer involving follicle depletion. Lancet Oncol. 2008, 9, 1108–1111. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.W.; Tyre, J.P.; Doherty, J.A.; Stram, D.A.; Kupryjanczyk, J.; Dansonka-Mieszkowska, A.; Plisiecka-Halasa, J.; Spiewankiewicz, B.; Myers, E.J.; Australian Cancer Study (Ovarian Cancer); et al. Evaluating the ovarian cancer gonadotropin hypothesis: A candidate gene study. Gynecol. Oncol. 2015, 136, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, J.R.; Patel, C.B.; Bapat, J.; Jones, C.M.; Ramos-Zapatero, M.; Ortell, K.K.; Tanios, R.; Haghighiabyaneh, M.; Axelrod, J.; DeStefano, J.W.; et al. Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet. 2020, 16, e1008558. [Google Scholar] [CrossRef]
- Nesic, K.; Wakefield, M.; Kondrashova, O.; Scott, C.L.; McNeish, I.A. Targeting DNA repair: The genome as a potential biomarker. J. Pathol. 2018, 244, 586–597. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Australian Ovarian Cancer Study Group; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- McGranahan, N.C. Swanton, Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Australian Ovarian Cancer Study Group; Davis, S.; D’Andrea, A.D.; Simpson, A.D.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.R.; Patel, C.B.; Willis, K.M.; Haghighiabyaneh, M.; Axelrod, J.; Tancioni, I.; Lu, D.; Bapat, J.; Young, S.; Cadassou, O.; et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat. Commun. 2017, 8, 14423. [Google Scholar] [CrossRef] [PubMed]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef]
- Chien, J.; Sicotte, H.; Fan, J.B.; Humphray, S.; Cunningham, J.M.; Kalli, K.R.; Oberg, A.L.; Hart, S.N.; Li, Y.; Davila, J.I.; et al. TP53 mutations, tetraploidy and homologous recombination repair defects in early stage high-grade serous ovarian cancer. Nucleic Acids Res. 2015, 43, 6945–6958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joruiz, S.M.; Bourdon, J.C. p53 Isoforms: Key regulators of the cell fate decision. Cold Spring Harb Perspect Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Kohler, M.F.; Marks, J.R.; Wiseman, R.W.; Jacobs, I.J.; Davidoff, A.M.; Clarke-Pearson, D.L.; Soper, J.T.; Bast, R.C., Jr.; Berchuck, A. Spectrum of mutation and frequency of allelic deletion of the p53 gene in ovarian cancer. J. Natl. Cancer Inst. 1993, 85, 1513–1519. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.-L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, A.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum and taxane-based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Mandilaras, V.; Garg, S.; Cabanero, M.; Tan, Q.; Pastrello, C.; Burnier, J.; Karakasis, K.; Wang, L.; Dhani, N.C.; Butler, M.O.; et al. TP53 mutations in high grade serous ovarian cancer and impact on clinical outcomes: A comparison of next generation sequencing and bioinformatics analyses. Int. J. Gynecol. Cancer 2019, 29, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Flippova, O.T.; Selenica, P.; Pareja, F.; Vahdatinia, M.; Zhu, Y.; Pei, X.; Riaz, N.; Roche, K.L.; Chi, D.S.; Abu-Rustum, N.R.; et al. Molecular characterization of high-grade serous ovarian cancers occurring in younger and older women. Gynecol. Oncol. 2021. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shumulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Schrader, K.A.; Hurlburt, J.; Kalloger, S.E.; Hansford, S.; Young, S.; Huntsman, D.G.; Gilks, B.; McAlpine, J.N. Germline BRCA1 and BRCA2 mutations in ovarian cancer: Utility of a histology-based referral strategy. Obstet Gynecol. 2012, 120, 235–240. [Google Scholar] [CrossRef]
- Pathania, S.; Bade, S.; Le Guillou, M.; Burke, K.; Reed, R.; Bowman-Colin, C.; Su, Y.; Ting, D.T.; Polyak, K.; Richardson, A.L.; et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun. 2014, 5, 5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, H.; Mohseni, M.; Tamaki, A.; Garay, J.P.; Croessmann, S.; Karnan, S.; Ota, A.; Wong, H.Y.; Konishi, Y.; Karakas, B.; et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17773–17778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca; Tp53; Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Austria, T.; Marion, C.; Yu, V.; Widschwendter, M.; Hinton, D.R.; Dubeau, L. Mechanism of cytokinesis failure in ovarian cystadenomas with defective BRCA1 and P53 pathways. Int. J. Cancer 2018, 143, 2932–2942. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Zhou, Q.; Iasonos, A.; Grisham, R.N.; Arnold, A.G.; Phillips, M.F.; Bhatia, J.; Levine, D.A.; Aghajanian, C.; Offit, K.; et al. Improved survival for BRCA2-associated serous ovarian cancer compared with both BRCA-negative and BRCA1-associated serous ovarian cancer. Cancer 2012, 118, 3703–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S.; et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef]
- Kuhn, E.; Wang, T.-L.; Doberstein, K.; Bahadirli-Talbott, A.; Ayhan, A.; Sehdev, A.S.; Drapkin, R.; Kurman, R.J.; Shih, I.-M. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: Further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod. Pathol. 2016, 29, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.M.; Enwere, E.; McIntyre, J.B.; Wilson, H.; Nwaroh, C.; Wiebe, N.; Ou, Y.; Liu, S.; Wiedemeyer, K.; Rambau, P.F.; et al. Combined CCNE1 high-level amplification and overexpression is associated with unfavourable outcome in tubo-ovarian high grade serous carcinoma. J. Pathol. Clin. Res. 2020, 6, 252–262. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Ferraresi, A.; Girone, C.; Esposito, A.; Vidoni, C.; Vallino, L.; Secomandi, E.; Dhanasekaran, D.N.; Isidoro, C. How Autophagy Shapes the Tumor Microenvironment in Ovarian Cancer. Front. Oncol. 2020, 10, 599915. [Google Scholar] [CrossRef]
- Ngabire, D.; Kim, G.D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef]
- Chui, M.H.; Wang, Y.; Wu, R.-C.; Seidman, J.; Kurman, R.J.; Wan, T.-L.; Shih, I.-M. Loss of ALDH1A1 expression is an early event in the pathogenesis of ovarian high-grade serous carcinoma. Mod. Pathol. 2015, 28, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Hou, Y.; Huang, Z.; Cai, J.; Wang, Z. SOX2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017, 108, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellner, K.; Miranda, F.; Fotso, D.C.; Herrero-Gonzalez, S.; Hayden, D.M.; Tearle, R.; Artibani, M.; KaramiNejadRanjbar, M.; Williams, R.; Gaitskell, K.; et al. Premalignant SOX2 overexpression in the fallopian tubes of ovarian cancer patients: Discovery and validation studies. EBioMedicine 2016, 10, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Boyd, C.; McCluggage, W.G. Low-Grade Ovarian Serous Neoplasms (Low-Grade Serous Carcinoma and Serous Borderline Tumor) Associated With High-Grade Serous Carcinoma or Undifferentiated Carcinoma: Report of a Series of Cases of an Unusual Phenomenon. Am. J. Surg. Pathol. 2012, 36, 368–375. [Google Scholar] [CrossRef]
- Emmanuel, C.; Chiew, Y.E.; George, J.; Etemadmoghadam, D.; Anglesio, M.S.; Sharma, R.; Russell, P.; Kennedy, C.; Fereday, S.; Hung, J.; et al. Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin. Cancer Res. 2014, 20, 6618–6630. [Google Scholar] [CrossRef] [Green Version]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih, I.e.M.; Kurman, R.J. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: A Rereview of cases lacking TP53 mutations in the cancer genome atlas ovarian study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsopoulos, J.; Lubinski, J.; Gronwald, J.; Cybulski, C.; Demsky, R.; Neuhausen, S.L.; Kim-Sing, C.; Tung, N.; Friedman, S.; Senter, L.; et al. Factors influencing ovulation and the risk of ovarian cancer in BRCA1 and BRCA2 mutation carriers. Int. J. Cancer 2015, 137, 1136–1146. [Google Scholar] [CrossRef] [Green Version]
- Trabert, B.; Ness, R.B.; Lo-Ciganic, W.H.; Murphy, M.A.; Goode, E.L.; Poole, E.M.; Brinton, L.A.; Webb, P.M.; Nagle, C.M.; Jordan, S.J.; et al. Aspirin, nonaspirin nonsteroidal antiinflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: A pooled analysis in the Ovarian Cancer Association Consortium. J. Natl. Cancer Inst. 2014, 106, djt431. [Google Scholar] [CrossRef] [Green Version]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattachaya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Wu, R.C.; Wang, P.; Lin, S.F.; Zhang, M.; Song, Q.; Chu, T.; Wang, B.G.; Kurman, R.J.; Vang, R.; Kinzler, K.; et al. Genomic landscape and evolutionary trajectories of ovarian cancer precursor lesions. J. Pathol. 2019, 248, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, I.; Kameda, S.; Hoshi, K. Early detection of ovarian and fallopian tube cancer by examination of cytological samples from the endometrial cavity. Br. J. Cancer 2013, 109, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Van Nagell, J.R., Jr.; Burgess, B.T.; Miller, R.W.; Baldwin, L.; DeSimone, C.P.; Ueland, E.R.; Huang, B.; Chen, Q.; Kryscio, R.J.; Pavlik, E.J. Survival of Women With Type I and II Epithelial Ovarian Cancer Detected by Ultrasound Screening. Obstet Gynecol. 2018, 132, 1091–1100. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; de Fazio, A.; Beroukhim, R.; Mermel, C.; George, J.; Getz, G.; Tothill, R.; Okamoto, A.; Raeder, M.B.; AOCS Study Group; et al. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin. Cancer Res. 2009, 15, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieser, V.; Gaugg, I.; Fleischer, M.; Shivalingaiah, G.; Wenzel, S.; Sprung, S.; Lax, S.F.; Zeimet, A.G.; Fiegl, H.; Marth, C. BRCA1/2 and TP53 mutation status associates with PD-1 and PD-L1 expression in ovarian cancer. Oncotarget 2018, 9, 17501–17511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Zhao, L.; Rojas, C.; Baterman, N.W.; Yao, H.; Lara, O.D.; Celestino, J.; Morgan, M.B.; Nguyen, T.V.; Conrads, K.A.; et al. Molecular Analysis of Clinically Defined Subsets of High-Grade Serous Ovarian Cancer. Cell Rep. 2020, 31, 107502. [Google Scholar] [CrossRef] [PubMed]
- Mittempergher, L. Genomic Characterization of High-Grade Serous Ovarian Cancer: Dissecting Its Molecular Heterogeneity as a Road Towards Effective Therapeutic Strategies. Curr. Oncol. Rep. 2016, 18, 44. [Google Scholar] [CrossRef]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [Green Version]
- Castellarin, M.; Milne, K.; Zeng, T.; Tse, K.; Mayo, M.; Zhao, Y.; Webb, J.R.; Watson, P.H.; Nelson, B.H.; Holt, R.A.; et al. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol. 2013, 229, 515–524. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsopoulos, J.B.; Rosen, I.; Fan, J.; Moody, J.R. McLaughlin, H.R.; Risch, H.; May, T.; Sun, P.; Narod, S.A. Ten-year survival after epithelial ovarian cancer is not associated with BRCA mutation status. Gynecol. Oncol. 2016, 140, 42–47. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otsuka, I. Mechanisms of High-Grade Serous Carcinogenesis in the Fallopian Tube and Ovary: Current Hypotheses, Etiologic Factors, and Molecular Alterations. Int. J. Mol. Sci. 2021, 22, 4409. https://doi.org/10.3390/ijms22094409
Otsuka I. Mechanisms of High-Grade Serous Carcinogenesis in the Fallopian Tube and Ovary: Current Hypotheses, Etiologic Factors, and Molecular Alterations. International Journal of Molecular Sciences. 2021; 22(9):4409. https://doi.org/10.3390/ijms22094409
Chicago/Turabian StyleOtsuka, Isao. 2021. "Mechanisms of High-Grade Serous Carcinogenesis in the Fallopian Tube and Ovary: Current Hypotheses, Etiologic Factors, and Molecular Alterations" International Journal of Molecular Sciences 22, no. 9: 4409. https://doi.org/10.3390/ijms22094409
APA StyleOtsuka, I. (2021). Mechanisms of High-Grade Serous Carcinogenesis in the Fallopian Tube and Ovary: Current Hypotheses, Etiologic Factors, and Molecular Alterations. International Journal of Molecular Sciences, 22(9), 4409. https://doi.org/10.3390/ijms22094409