
Characterization of the Survival Influential Genes in Carcinogenesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
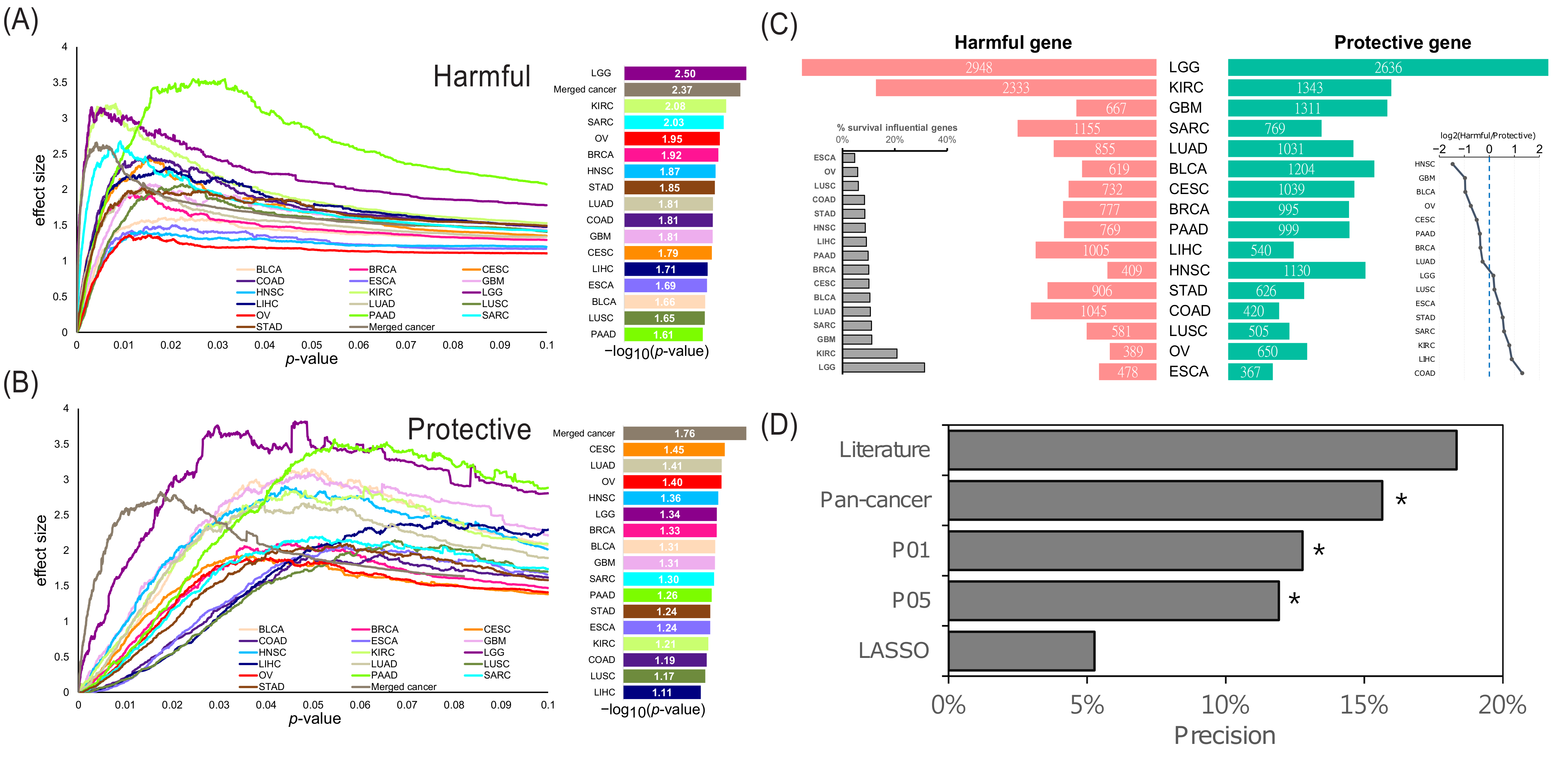
2.1. Overview of the Identified Survival Influential Genes in Cancers
2.2. Exclusivity of the SIGs and Identification of the Pan-Cancer SIGs
2.3. Analysis of SIG Roles in the Human Co-Expressed Protein Interaction Network
2.4. The Survival Influential Functional Modules
2.5. The Cancer Hallmarks of SIGs in Pan-Cancer
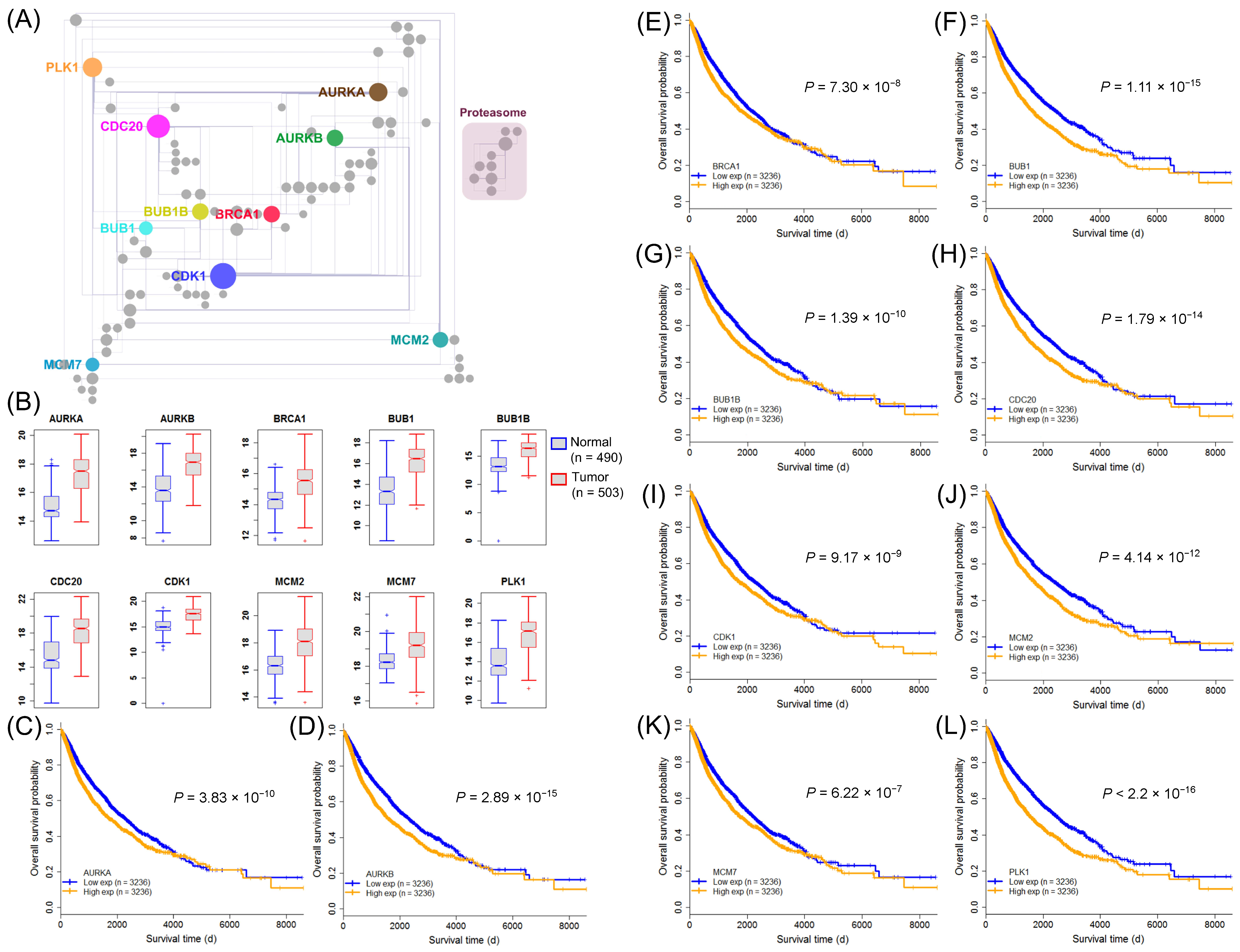
2.6. Identification of Clinically Relevant Pan-Cancer Harmful SIGs in the Proliferation Hallmark
3. Discussion
4. Materials and Methods
4.1. Data Collection and Preprocessing
4.2. Identification of Survival Influential Genes in Cancers and Pan-Cancer
4.3. Compilation of the Cancer-Associated Genes
4.4. Functional Modules of SIGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerdes, S.; Edwards, R.; Kubal, M.; Fonstein, M.; Stevens, R.; Osterman, A. Essential genes on metabolic maps. Curr. Opin. Biotechnol. 2006, 17, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; Eberl, L.; Glass, J.I. Essence of life: Essential genes of minimal genomes. Trends Cell Biol. 2011, 21, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, A.M.; Snitkin, E.S.; Parker, S.C.; DeLisi, C.; Kasif, S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genom. 2006, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Acencio, M.L.; Lemke, N. Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinform. 2009, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Lin, C.Y.; Chang, S.L.; Chao, T.F.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Hu, Y.F.; Tuan, T.C.; Liao, J.N.; et al. Risk of Stroke in Patients With Short-Run Atrial Tachyarrhythmia. Stroke 2017, 48, 3232–3238. [Google Scholar] [CrossRef]
- Kleinbaum, D.G.; Klein, M. Survival Analysis: A Self-Learning Text, 3rd ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [Green Version]
- Butland, G.; Babu, M.; Diaz-Mejia, J.J.; Bohdana, F.; Phanse, S.; Gold, B.; Yang, W.; Li, J.; Gagarinova, A.G.; Pogoutse, O.; et al. eSGA: E. coli synthetic genetic array analysis. Nat. Methods 2008, 5, 789–795. [Google Scholar] [CrossRef]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef]
- Glass, J.I.; Assad-Garcia, N.; Alperovich, N.; Yooseph, S.; Lewis, M.R.; Maruf, M.; Hutchison, C.A.; Smith, H.O.; Venter, J.C. Essential Genes of a Minimal Bacterium. Proc. Natl. Acad. Sci. USA 2006, 103, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Anaya, J.; Reon, B.; Chen, W.M.; Bekiranov, S.; Dutta, A. A pan-cancer analysis of prognostic genes. PeerJ 2015, 3, e1499. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Li, Y.; Liu, C.J.; Xiang, Y.; Li, C.; Ye, Y.; Zhang, Z.; Hawke, D.H.; Park, P.K.; Diao, L.; et al. A Pan-cancer Analysis of the Expression and Clinical Relevance of Small Nucleolar RNAs in Human Cancer. Cell Rep. 2017, 21, 1968–1981. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhao, Z. A comparative study of cancer proteins in the human protein-protein interaction network. BMC Genom. 2010, 11 (Suppl. 3), S5. [Google Scholar] [CrossRef] [Green Version]
- Taylor, I.W.; Linding, R.; Warde-Farley, D.; Liu, Y.; Pesquita, C.; Faria, D.; Bull, S.; Pawson, T.; Morris, Q.; Wrana, J.L. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 2009, 27, 199–204. [Google Scholar] [CrossRef]
- Li, T.; Wernersson, R.; Hansen, R.B.; Horn, H.; Mercer, J.; Slodkowicz, G.; Workman, C.T.; Rigina, O.; Rapacki, K.; Staerfeldt, H.H.; et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat. Methods 2017, 14, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.; Jacks, T.; Pavletich, N.P. The cell cycle and cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 2776–2778. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Adam, J.K.; Odhav, B.; Bhoola, K.D. Immune responses in cancer. Pharmacol. Ther. 2003, 99, 113–132. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.W.; Pan, Y.; Tsai, B.L.; Xie, Z.; Demirdjian, L.; Xiao, W.; Yang, H.T.; Zhang, Y.; Lin, C.H.; Cheng, D.; et al. Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc. Natl. Acad. Sci. USA 2020, 117, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16 (Suppl. 1), S87–S105. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, G.; Sengupta, S.; Panda, C.K.; Gollin, S.M.; Saunders, W.S.; Roychoudhury, S. Overexpression of Cdc20 leads to impairment of the spindle assembly checkpoint and aneuploidization in oral cancer. Carcinogenesis 2007, 28, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Kim, Y.; Lee, J.H.; Kim, Y.S. High expression of spindle assembly checkpoint proteins CDC20 and MAD2 is associated with poor prognosis in urothelial bladder cancer. Virchows Arch. Int. J. Pathol. 2013, 463, 681–687. [Google Scholar] [CrossRef]
- Chang, D.Z.; Ma, Y.; Ji, B.; Liu, Y.; Hwu, P.; Abbruzzese, J.L.; Logsdon, C.; Wang, H. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J. Hematol. Oncol. 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.J.; Hu, K.S.; Wang, D.S.; Zeng, Z.L.; Zhang, D.S.; Chen, D.L.; Bai, L.; Xu, R.H. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J. Transl. Med. 2013, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Karra, H.; Repo, H.; Ahonen, I.; Loyttyniemi, E.; Pitkanen, R.; Lintunen, M.; Kuopio, T.; Soderstrom, M.; Kronqvist, P. Cdc20 and securin overexpression predict short-term breast cancer survival. Br. J. Cancer 2014, 110, 2905–2913. [Google Scholar] [CrossRef] [Green Version]
- Mao, D.D.; Gujar, A.D.; Mahlokozera, T.; Chen, I.; Pan, Y.; Luo, J.; Brost, T.; Thompson, E.A.; Turski, A.; Leuthardt, E.C.; et al. A CDC20-APC/SOX2 Signaling Axis Regulates Human Glioblastoma Stem-like Cells. Cell Rep. 2015, 11, 1809–1821. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Daigo, Y.; Aragaki, M.; Ishikawa, K.; Sato, M.; Kaji, M. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J. Surg. Oncol. 2012, 106, 423–430. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef] [Green Version]
- Tzafrir, I.; Pena-Muralla, R.; Dickerman, A.; Berg, M.; Rogers, R.; Hutchens, S.; Sweeney, T.C.; McElver, J.; Aux, G.; Patton, D.; et al. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004, 135, 1206–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, Z.; Chen, W.; Shang, J.; Li, X.; Nie, Z.; Yang, J.; Li, N. Clinicopathological and prognostic implications of polo-like kinase 1 expression in colorectal cancer: A systematic review and meta-analysis. Gene 2019, 721, 144097. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.; Sun, Z.; Guo, Q.; Shi, H.; Jia, Y. The clinical and prognostic value of polo-like kinase 1 in lung squamous cell carcinoma patients: Immunohistochemical analysis. Biosci. Rep. 2017, 37, BSR20170852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; Kamath, A.; McDade, F.; Rogers, D.; Brooks, A.N.; Zhu, J.; Haussler, D. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019, 326470. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model, 1st ed.; Springer: New York, NY, USA, 2000. [Google Scholar]
- Andersen, P.K.; Gill, R.D. Cox’s Regression Model for Counting Processes: A Large Sample Study. Ann. Stat. 1982, 10, 1100–1120. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in S. Version 2.38. 2015. Available online: https://CRAN.R-project.org/package=survival (accessed on 16 October 2016).
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Kim, S.Y.; Kim, C.W.; Roh, S.A.; Ha, Y.J.; Lee, J.L.; Heo, H.; Cho, D.H.; Lee, J.S.; Kim, Y.S.; et al. A prognostic index based on an eleven gene signature to predict systemic recurrences in colorectal cancer. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Hou, Z.; Yang, J.; Wang, H.; Liu, D.; Zhang, H. A Potential Prognostic Gene Signature for Predicting Survival for Glioblastoma Patients. BioMed Res. Int. 2019, 2019, 9506461. [Google Scholar] [CrossRef] [Green Version]
- Haider, S.; Wang, J.; Nagano, A.; Desai, A.; Arumugam, P.; Dumartin, L.; Fitzgibbon, J.; Hagemann, T.; Marshall, J.F.; Kocher, H.M.; et al. A multi-gene signature predicts outcome in patients with pancreatic ductal adenocarcinoma. Genome Med. 2014, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.J.; Rees, J.R.; Hardwick, R.H.; Hardwick, J.S.; Vowler, S.L.; Ong, C.A.; Zhang, C.; Save, V.; O’Donovan, M.; Rassl, D.; et al. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 2010, 139, 1995–2004.e15. [Google Scholar] [CrossRef] [Green Version]
- Van de Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Boutros, P.C.; Lau, S.K.; Pintilie, M.; Liu, N.; Shepherd, F.A.; Der, S.D.; Tsao, M.S.; Penn, L.Z.; Jurisica, I. Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 2824–2828. [Google Scholar] [CrossRef] [Green Version]
- Consortium, T.G.O. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Hsiang, J.T.; Wu, C.Y.; Oyang, Y.J.; Juan, H.F.; Huang, H.C. Dynamic functional modules in co-expressed protein interaction networks of dilated cardiomyopathy. BMC Syst. Biol. 2010, 4, 138. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Schlicker, A.; Domingues, F.S.; Rahnenfuhrer, J.; Lengauer, T. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinform. 2006, 7, 302. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, I.; Lugmayr, A.; Siira, S.J.; Rackham, O.; Filipovska, A. CirGO: An alternative circular way of visualising gene ontology terms. BMC Bioinform. 2019, 20, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, D.; Chang, Y.-L.; Lin, Y.-C.; Lin, C.-C. Characterization of the Survival Influential Genes in Carcinogenesis. Int. J. Mol. Sci. 2021, 22, 4384. https://doi.org/10.3390/ijms22094384
Sahu D, Chang Y-L, Lin Y-C, Lin C-C. Characterization of the Survival Influential Genes in Carcinogenesis. International Journal of Molecular Sciences. 2021; 22(9):4384. https://doi.org/10.3390/ijms22094384
Chicago/Turabian StyleSahu, Divya, Yu-Lin Chang, Yin-Chen Lin, and Chen-Ching Lin. 2021. "Characterization of the Survival Influential Genes in Carcinogenesis" International Journal of Molecular Sciences 22, no. 9: 4384. https://doi.org/10.3390/ijms22094384
APA StyleSahu, D., Chang, Y. -L., Lin, Y. -C., & Lin, C. -C. (2021). Characterization of the Survival Influential Genes in Carcinogenesis. International Journal of Molecular Sciences, 22(9), 4384. https://doi.org/10.3390/ijms22094384