
Computational-Driven Epitope Verification and Affinity Maturation of TLR4-Targeting Antibodies


Abstract
:1. Introduction
2. Results
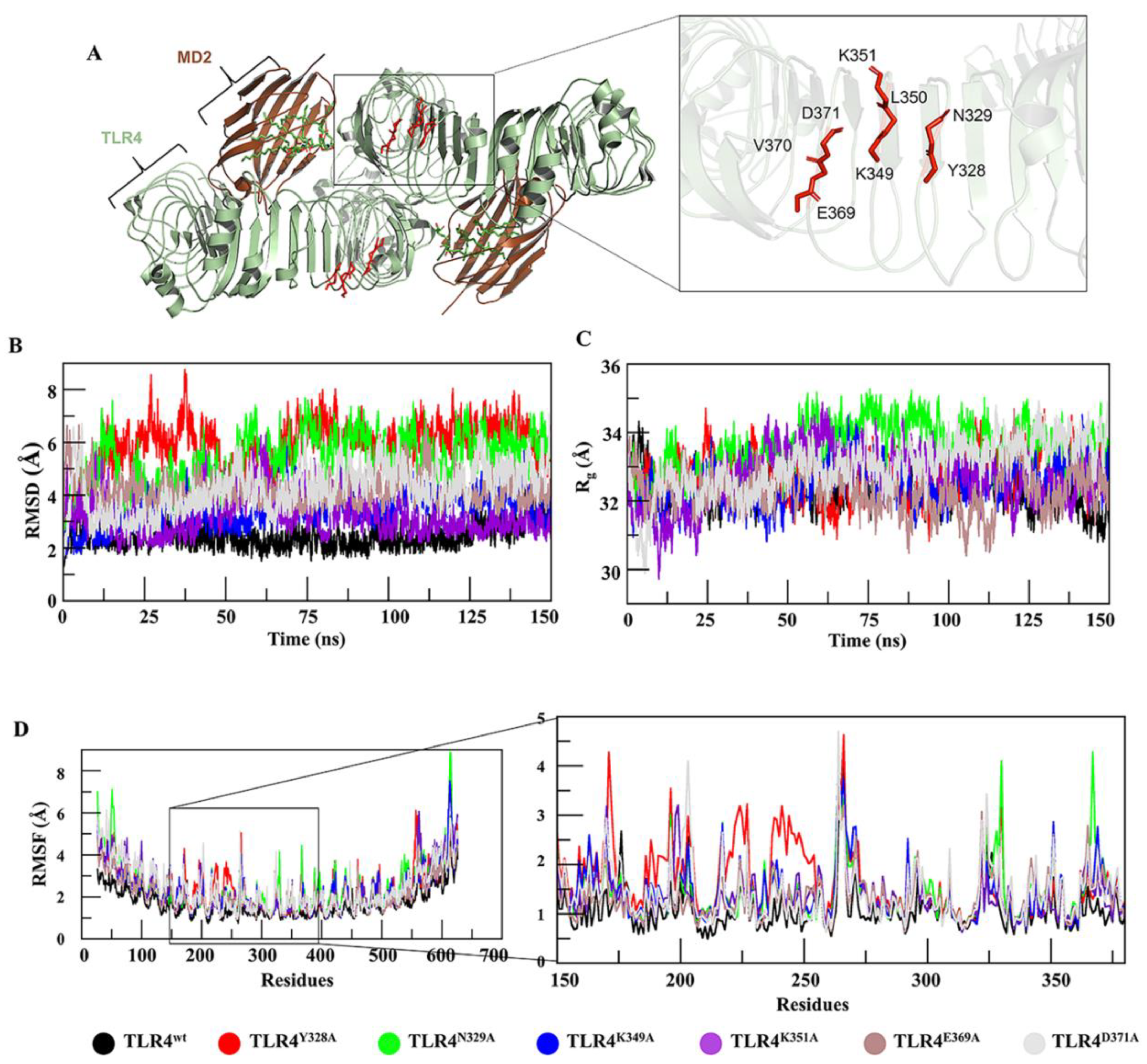
2.1. Computational TLR4 Epitope Mutagenesis and Network Analysis
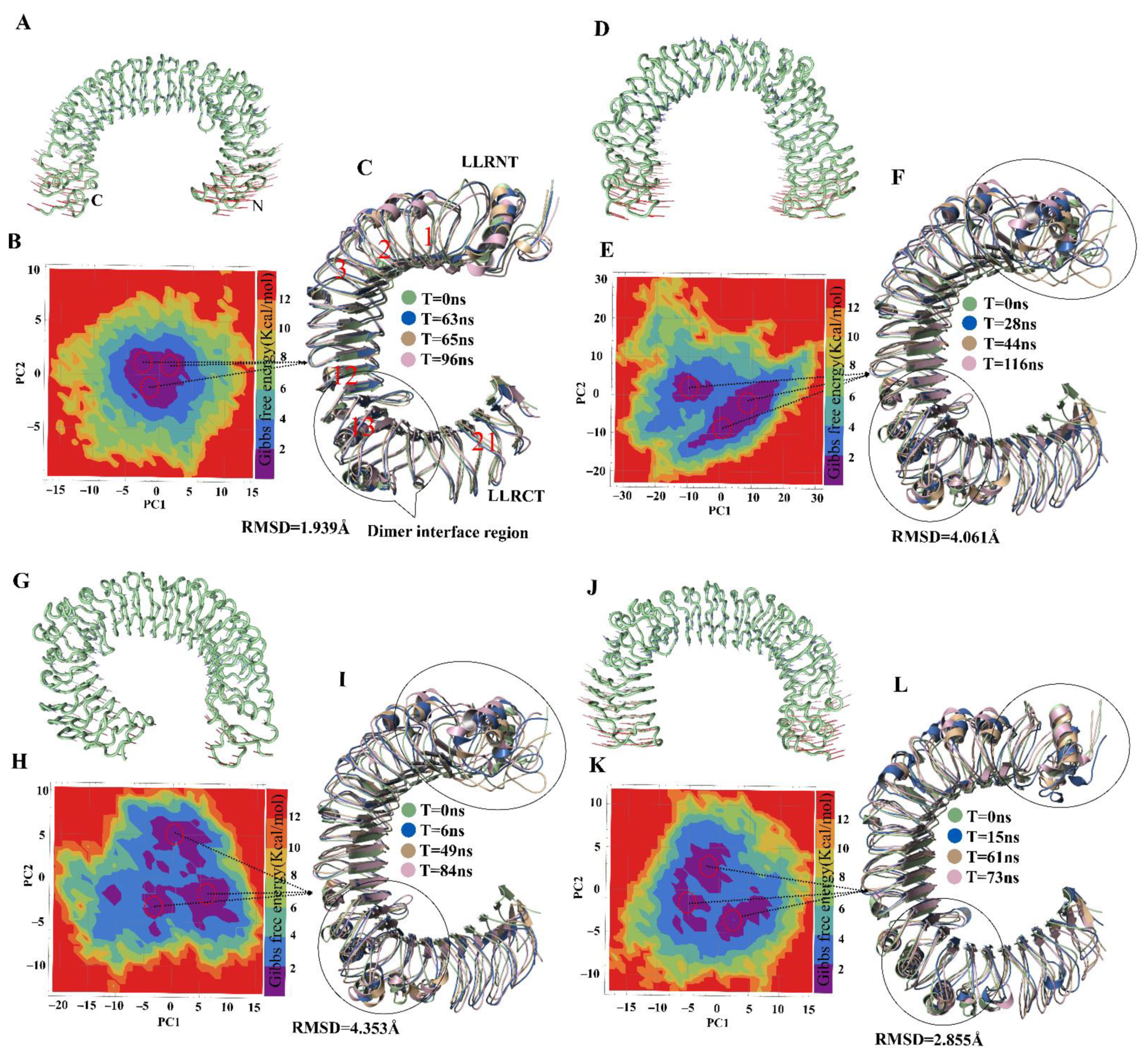
2.2. MD of TLR4wt and Muteins
2.3. Mutation-Induced Conformational Changes in TLR4 Structure
2.4. Visualization and Identification of Principal Motion of TLR4wt and Muteins
2.5. Energetics of Conformation Transitions
2.6. The Influence of Mutations on the TLR4-mAb Interaction and Binding Affinity
2.7. Epitope Prediction and Interaction Analysis of the Epitope–Paratope Interface for Rational Design of Antibodies
3. Discussion
4. Materials and Methods
4.1. In Silico Structure Reconstruction and Mutation of TLR4
4.2. Construction of the Interaction Network
4.3. Epitope Prediction and CDR Annotations
4.4. Molecular Docking and MD Simulation
4.5. Binding-Free-Energy Calculations
4.6. Principal Component Analysis and Free-Energy Landscape
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Bryant, C.E.; Doyle, S.L. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol. Rev. 2009, 61, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Kim, G.Y.; Achek, A.; Cho, E.Y.; Baek, W.Y.; Choi, Y.S.; Lee, W.H.; Kim, D.J.; Lee, S.H.; Kim, W.; et al. The alphaC helix of TIRAP holds therapeutic potential in TLR-mediated autoimmune diseases. Biomaterials 2020, 245, 119974. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yan, S.; Wang, H.; Gu, B.; Sun, K.; Yang, X.; Sun, B.; Wang, X. IL-29 Enhances LPS/TLR4-Mediated Inflammation in Rheumatoid Arthritis. Cell. Physiol. Biochem. 2015, 37, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Achek, A.; Shah, M.; Seo, J.Y.; Kwon, H.K.; Gui, X.; Shin, H.J.; Cho, E.Y.; Lee, B.S.; Kim, D.J.; Lee, S.H.; et al. Linear and Rationally Designed Stapled Peptides Abrogate TLR4 Pathway and Relieve Inflammatory Symptoms in Rheumatoid Arthritis Rat Model. J. Med. Chem. 2019, 62, 6495–6511. [Google Scholar] [CrossRef]
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. 2016, 7, e2234. [Google Scholar] [CrossRef]
- Yao, L.; Kan, E.M.; Lu, J.; Hao, A.; Dheen, S.T.; Kaur, C.; Ling, E.A. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: Role of TLR4 in hypoxic microglia. J. Neuroinflammation 2013, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Teng, W.; Wang, L.; Xue, W.; Guan, C. Activation of TLR4-mediated NFkappaB signaling in hemorrhagic brain in rats. Mediat. Inflamm. 2009, 2009, 473276. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haabeth, O.A.; Bogen, B.; Corthay, A. A model for cancer-suppressive inflammation. Oncoimmunology 2012, 1, 1146–1155. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Bruce, N.J.; Ganotra, G.K.; Kokh, D.B.; Sadiq, S.K.; Wade, R.C. New approaches for computing ligand-receptor binding kinetics. Curr. Opin. Struct. Biol. 2018, 49, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.A.; Ambrosetti, F.; Bonvin, A.; Colwell, L.J.; Kelm, S.; Kumar, S.; Krawczyk, K. Computational approaches to therapeutic antibody design: Established methods and emerging trends. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T. Toward rational antibody design: Recent advancements in molecular dynamics simulations. Int. Immunol. 2018, 30, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zeng, H.; Mueller, J.; Carter, B.; Wang, Z.; Schilz, J.; Horny, G.; Birnbaum, M.E.; Ewert, S.; Gifford, D.K. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 2020, 36, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Friedensohn, S.; Neumeier, D.; Khan, T.A.; Csepregi, L.; Parola, C.; de Vries, A.R.G.; Erlach, L.; Mason, D.M.; Reddy, S.T. Convergent selection in antibody repertoires is revealed by deep learning. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hatterer, E.; Shang, L.; Simonet, P.; Herren, S.; Daubeuf, B.; Teixeira, S.; Reilly, J.; Elson, G.; Nelson, R.; Gabay, C.; et al. A specific anti-citrullinated protein antibody profile identifies a group of rheumatoid arthritis patients with a toll-like receptor 4-mediated disease. Arthritis Res. Ther. 2016, 18, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn-Siegrist, I.; Leger, O.; Daubeuf, B.; Poitevin, Y.; Depis, F.; Herren, S.; Kosco-Vilbois, M.; Dean, Y.; Pugin, J.; Elson, G. Pivotal involvement of Fcgamma receptor IIA in the neutralization of lipopolysaccharide signaling via a potent novel anti-TLR4 monoclonal antibody 15C1. J. Biol. Chem. 2007, 282, 34817–34827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, L.; Daubeuf, B.; Triantafilou, M.; Olden, R.; Depis, F.; Raby, A.C.; Herren, S.; Dos Santos, A.; Malinge, P.; Dunn-Siegrist, I.; et al. Selective antibody intervention of Toll-like receptor 4 activation through Fc gamma receptor tethering. J. Biol. Chem. 2014, 289, 15309–15318. [Google Scholar] [CrossRef] [Green Version]
- Monnet, E.; Lapeyre, G.; Poelgeest, E.V.; Jacqmin, P.; Graaf, K.; Reijers, J.; Moerland, M.; Burggraaf, J.; Min, C. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin. Pharmacol. Ther. 2017, 101, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Monnet, E.; Choy, E.H.; McInnes, I.; Kobakhidze, T.; de Graaf, K.; Jacqmin, P.; Lapeyre, G.; de Min, C. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: A phase II study. Ann. Rheum. Dis. 2020, 79, 316–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyau, J.; Didelot, G.; Malinge, P.; Ravn, U.; Magistrelli, G.; Depoisier, J.F.; Pontini, G.; Poitevin, Y.; Kosco-Vilbois, M.; Fischer, N.; et al. Robust Antibody-Antigen Complexes Prediction Generated by Combining Sequence Analyses, Mutagenesis, In Vitro Evolution, X-ray Crystallography and In Silico Docking. J. Mol. Biol. 2015, 427, 2647–2662. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Yamagata, Y.; Ukai, I.; Takeuchi, S.; Okubo, M.; Kobayashi, Y.; Kozakai, S.; Kubota, K.; Numasaki, M.; Kanemitsu, Y.; et al. An inhibitory epitope of human Toll-like receptor 4 resides on leucine-rich repeat 13 and is recognized by a monoclonal antibody. FEBS Lett. 2017, 591, 2406–2416. [Google Scholar] [CrossRef] [Green Version]
- Kamenik, A.S.; Handle, P.H.; Hofer, F.; Kahler, U.; Kraml, J.; Liedl, K.R. Polarizable and non-polarizable force fields: Protein folding, unfolding, and misfolding. J. Chem. Phys. 2020, 153, 185102. [Google Scholar] [CrossRef]
- Jana, K.; Kepp, K.P. Force-Field Benchmarking by Alternatives: A Systematic Study of Ten Small α- and β-Proteins. bioRxiv 2020, 974477. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Chuah, C.; Majeed, A.B.A.; Leow, C.H.; Lim, B.H.; Leow, C.Y. Current progress of immunoinformatics approach harnessed for cellular- and antibody-dependent vaccine design. Pathog. Glob. Health 2018, 112, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Araya, C.L.; Fleishman, S.J.; Kellogg, E.H.; Stephany, J.J.; Baker, D.; Fields, S. High-resolution mapping of protein sequence-function relationships. Nat. Methods 2010, 7, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.A.; Dessailly, B.H.; Orengo, C.A. Residue mutations and their impact on protein structure and function: Detecting beneficial and pathogenic changes. Biochem. J. 2013, 449, 581–594. [Google Scholar] [CrossRef]
- Schaefer, C.; Rost, B. Predict impact of single amino acid change upon protein structure. BMC Genomics 2012, 13 (Suppl. 4), S4. [Google Scholar] [CrossRef] [Green Version]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Mizohata, E.; Nagatoishi, S.; Watanabe, T.; Nakakido, M.; Iwanari, H.; Mochizuki, Y.; Nakayama, T.; Kado, Y.; Yokota, Y.; et al. Affinity Improvement of a Cancer-Targeted Antibody through Alanine-Induced Adjustment of Antigen-Antibody Interface. Structure 2019, 27, 519–527.e515. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Wong, L.; Lu, L.; Hoi, S.C.; Li, J. B-cell epitope prediction through a graph model. BMC Bioinform. 2012, 13 (Suppl. 17), S20. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. Chembiochem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Loyau, J.; Malinge, P.; Daubeuf, B.; Shang, L.; Elson, G.; Kosco-Vilbois, M.; Fischer, N.; Rousseau, F. Maximizing the potency of an anti-TLR4 monoclonal antibody by exploiting proximity to Fcgamma receptors. MAbs 2014, 6, 1621–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Ghosh, N.; Slivka, P.F.; Fiorini, Z.; Hutchinson, M.R.; Watkins, L.R.; Yin, H. An MD2 hot-spot-mimicking peptide that suppresses TLR4-mediated inflammatory response in vitro and in vivo. Chembiochem 2011, 12, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, C.; Mitsuzawa, H.; Sano, H.; Shimizu, T.; Matsushima, N.; Kuroki, Y. Toll-like receptor 4 region Glu24-Lys47 is a site for MD-2 binding: Importance of CYS29 and CYS40. J. Biol. Chem. 2006, 281, 38322–38329. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, C.; Mitsuzawa, H.; Hyakushima, N.; Sano, H.; Matsushima, N.; Kuroki, Y. The Toll-like receptor 4 region Glu24-Pro34 is critical for interaction with MD-2. Biochem. Biophys. Res. Commun. 2005, 328, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Re, F.; Strominger, J.L. Separate functional domains of human MD-2 mediate Toll-like receptor 4-binding and lipopolysaccharide responsiveness. J. Immunol. 2003, 171, 5272–5276. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, R.M.; Srinivasan, N. Stability of domain structures in multi-domain proteins. Sci. Rep. 2011, 1, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brysbaert, G.; Mauri, T.; de Ruyck, J.; Lensink, M.F. Identification of Key Residues in Proteins Through Centrality Analysis and Flexibility Prediction with RINspector. Curr. Protoc. Bioinform. 2019, 65, e66. [Google Scholar] [CrossRef] [Green Version]
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Marks, C.; Nowak, J.; Regep, C.; Georges, G.; Kelm, S.; Popovic, B.; Deane, C.M. SAbPred: A structure-based antibody prediction server. Nucleic Acids Res. 2016, 44, W474–W478. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, K.; Liu, X.; Baker, T.; Shi, J.; Deane, C.M. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 2014, 30, 2288–2294. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Ritleng, P.; Loubiere, R.; Marcelet, B. [Suppurative pseudo-lithiasic canaliculitis]. Ophtalmologie 1989, 3, 1–3. [Google Scholar] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Skjaerven, L.; Yao, X.Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Linssen, A.B.; de Groot, B.L.; van Aalten, D.M.; Berendsen, H.J. An efficient method for sampling the essential subspace of proteins. J. Biomol. Struct. Dyn. 1996, 13, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex-Type | Vdw Energy | Electrostatic Energy | Polar Solvation | SASA | Binding Energy |
|---|---|---|---|---|---|
| Hu 15C1-Ctl | −379.741 +/− 18.392 | −3222.642 +/− 100.808 | 776.278 +/− 54.509 | −42.161 +/− 3.383 | −2868.266 +/− 98.367 |
| C2E3-Ctl | −458.220 +/− 22.391 | −3339.736 +/− 98.973 | 1059.671 +/− 65.271 | −58.917 +/− 4.214 | −2797.202 +/− 97.581 |
| Hu 15C1-Y328A | −341.401 +/− 20.569 | −3232.006 +/− 172.050 | 1139.37 +/− 100.126 | −56.347 +/− 3.599 | −2490.384 +/− 169.412 |
| Hu 15C1-N329A | −330.692 +/− 20.298 | −3360.506 +/− 84.052 | 1377.683 +/− 124.152 | −63.289 +/− 3.401 | −2376.804 +/− 131.607 |
| Hu 15C1-K349A | −336.232 +/− 20.298 | −3260.506 +/− 84.052 | 1377.673 +/− 124.152 | −55.389 +/− 3.401 | −2274.454 +/− 131.607 |
| Hu 15C1-EP1 | −514.827 +/− 27.258 | −4178.843 +/− 126.407 | 1398.977 +/− 85.365 | −63.568 +/− 3.580 | −3358.261 +/− 96.088 |
| C2E3-EP1 | −619.706 +/− 28.893 | −6359.128 +/− 190.555 | 1996.806 +/− 109.523 | −86.952 +/− 3.889 | −5068.979 +/− 115.771 |
| Hu 15C1-EP2 | −385.218 +/− 19.617 | −2301.460 +/− 71.852 | 1004.632 +/− 113.944 | −48.778 +/− 3.287 | −1730.824 +/− 122.321 |
| C2E3-EP2 | −344.892 +/− 19.082 | −3348.181 +/− 107.983 | 885.182 +/− 71.196 | −41.990 +/− 3.310 | −2849.881 +/− 61.910 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, B.; Batool, M.; Kim, M.-S.; Choi, S. Computational-Driven Epitope Verification and Affinity Maturation of TLR4-Targeting Antibodies. Int. J. Mol. Sci. 2021, 22, 5989. https://doi.org/10.3390/ijms22115989
Ahmad B, Batool M, Kim M-S, Choi S. Computational-Driven Epitope Verification and Affinity Maturation of TLR4-Targeting Antibodies. International Journal of Molecular Sciences. 2021; 22(11):5989. https://doi.org/10.3390/ijms22115989
Chicago/Turabian StyleAhmad, Bilal, Maria Batool, Moon-Suk Kim, and Sangdun Choi. 2021. "Computational-Driven Epitope Verification and Affinity Maturation of TLR4-Targeting Antibodies" International Journal of Molecular Sciences 22, no. 11: 5989. https://doi.org/10.3390/ijms22115989
APA StyleAhmad, B., Batool, M., Kim, M. -S., & Choi, S. (2021). Computational-Driven Epitope Verification and Affinity Maturation of TLR4-Targeting Antibodies. International Journal of Molecular Sciences, 22(11), 5989. https://doi.org/10.3390/ijms22115989