
Recent Developments on gMicroMC: Transport Simulations of Proton and Heavy Ions and Concurrent Transport of Radicals and DNA



Abstract
:1. Introduction
2. Materials and Methods
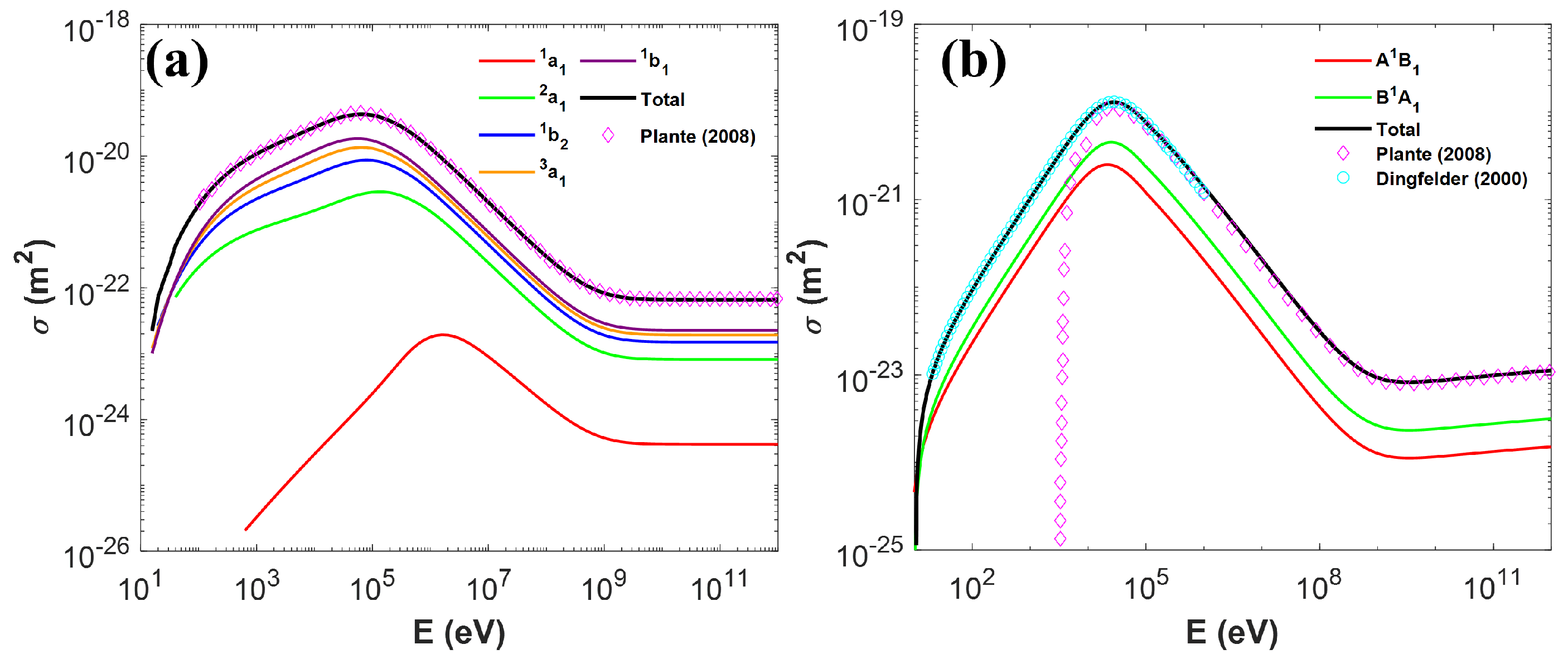
2.1. Cross-Sections for the Transport Simulation of Protons and Heavy Ions
2.1.1. Ionization for Protons
2.1.2. Excitation for Proton
2.1.3. Charge Effect
2.1.4. Cross-Section for Heavy Ions
2.2. Concurrent Transport Method
2.3. GPU Implementation
2.3.1. Physical Transport for Protons and Heavy Ions
2.3.2. Concurrent Transport
2.4. Simulation Setup
2.4.1. Simulation Setup for the Transport of Protons and Heavy Ions
2.4.2. Simulation Setup for Concurrent Transport
3. Results
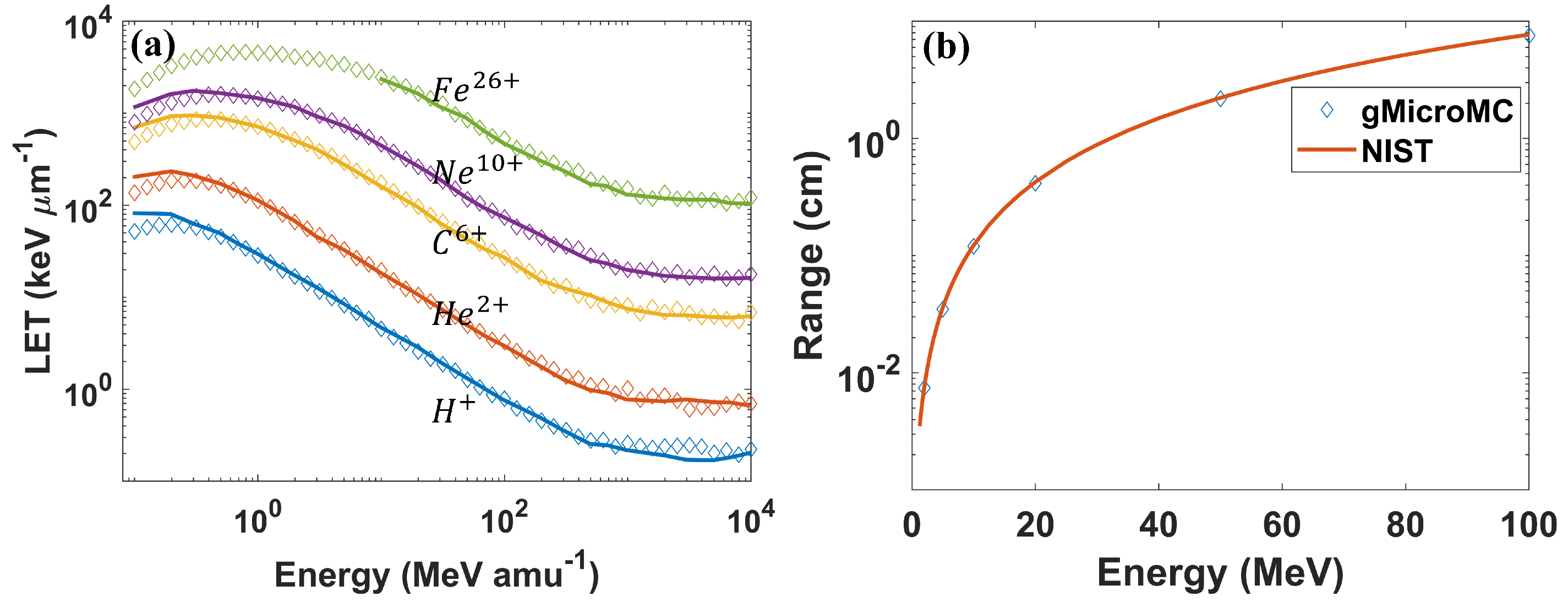
3.1. Validation of Development for Protons and Heavy Ions
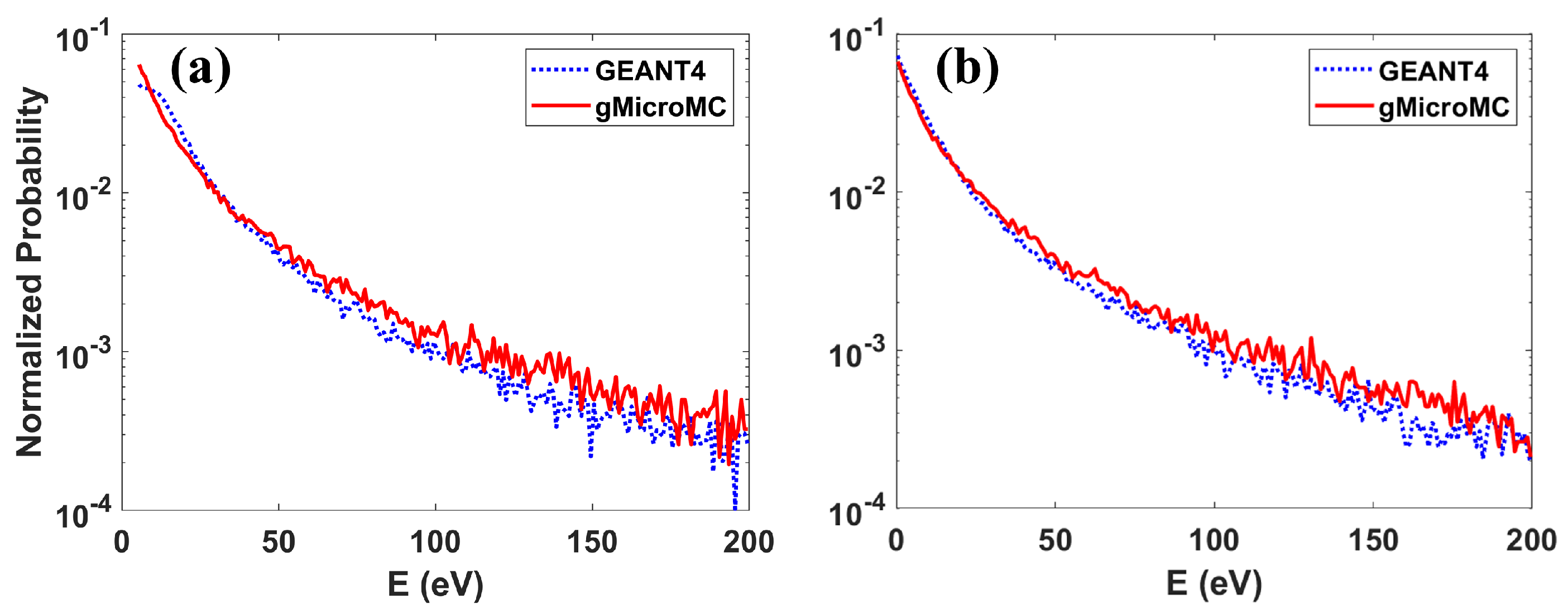
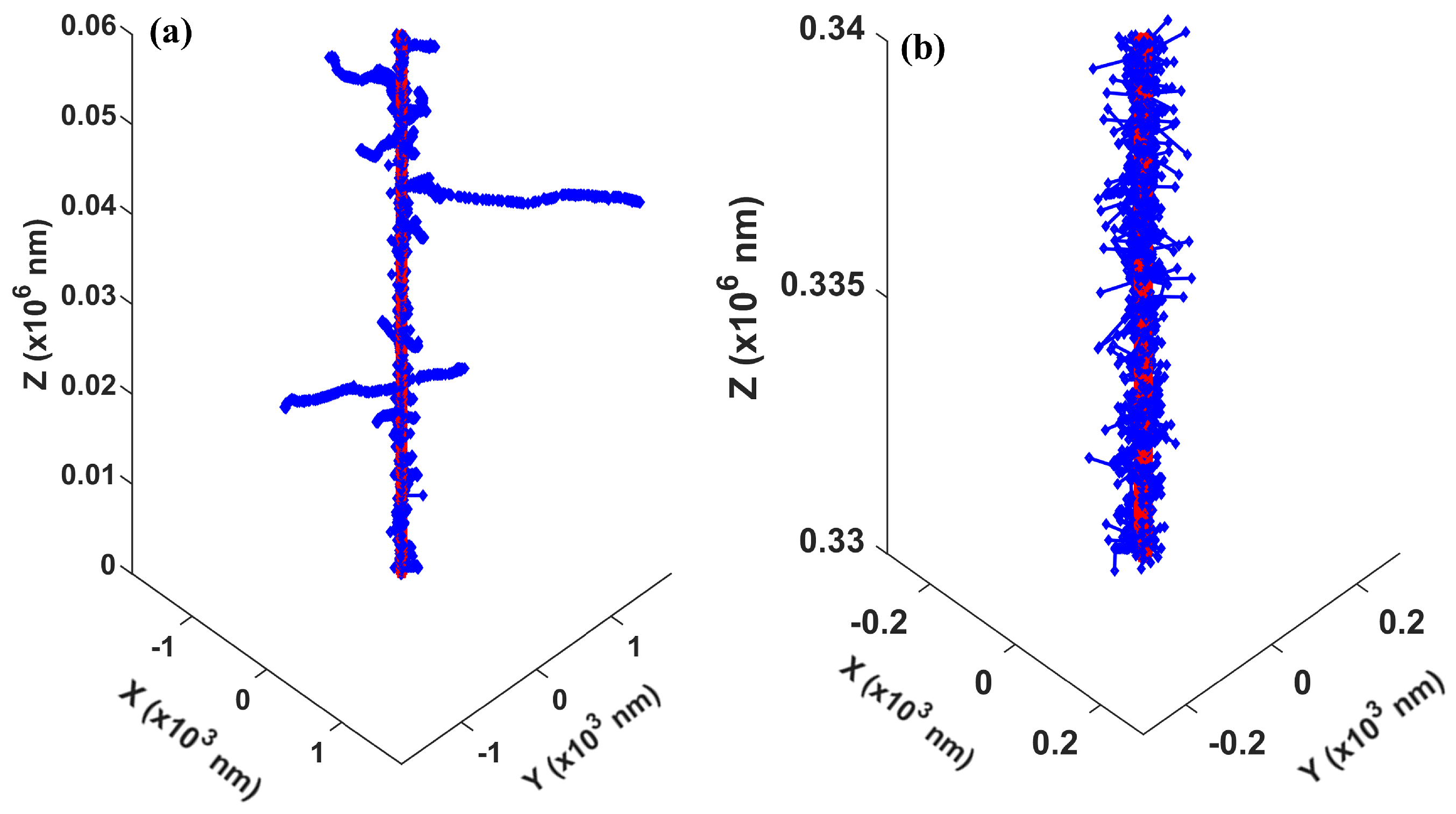
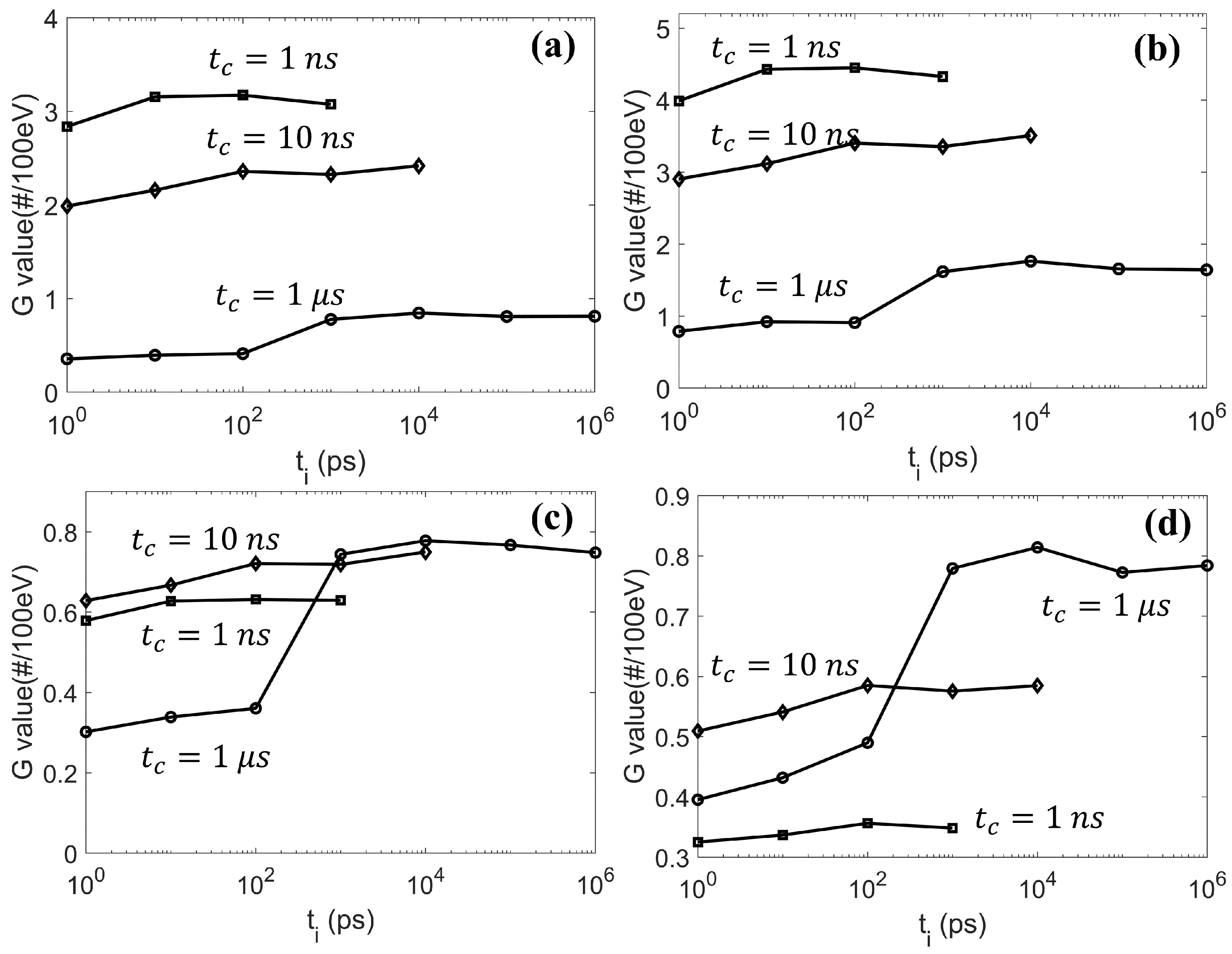
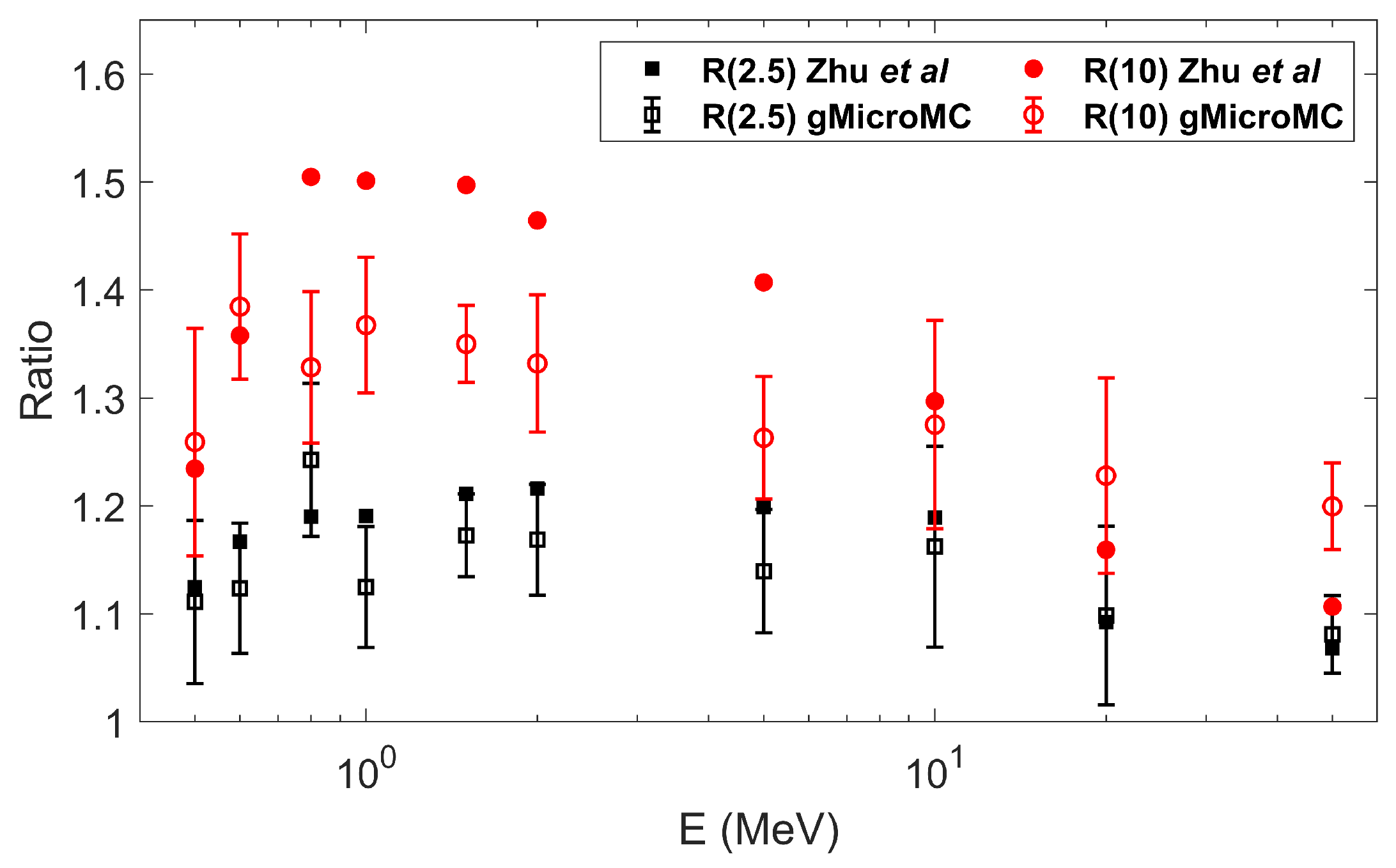
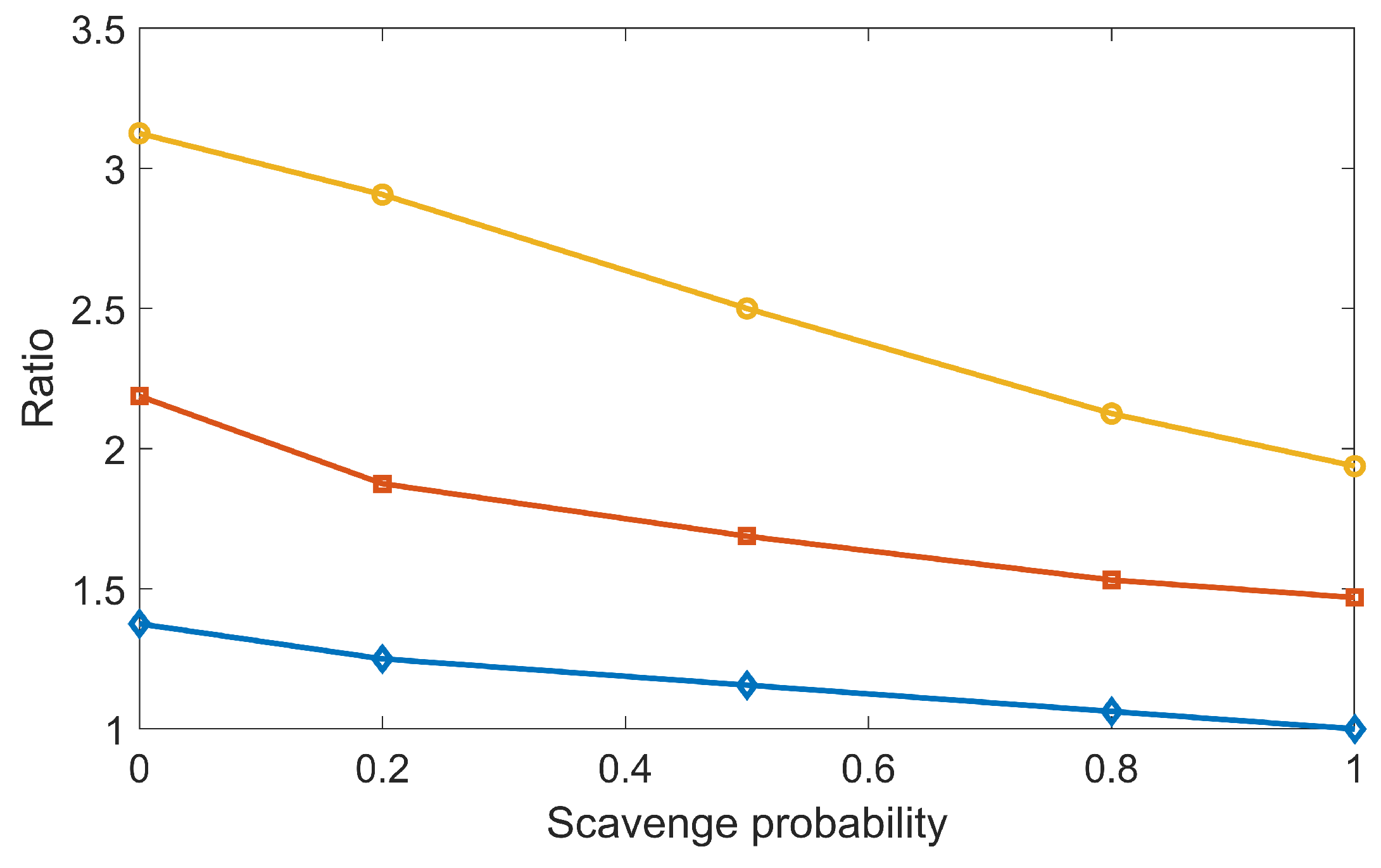
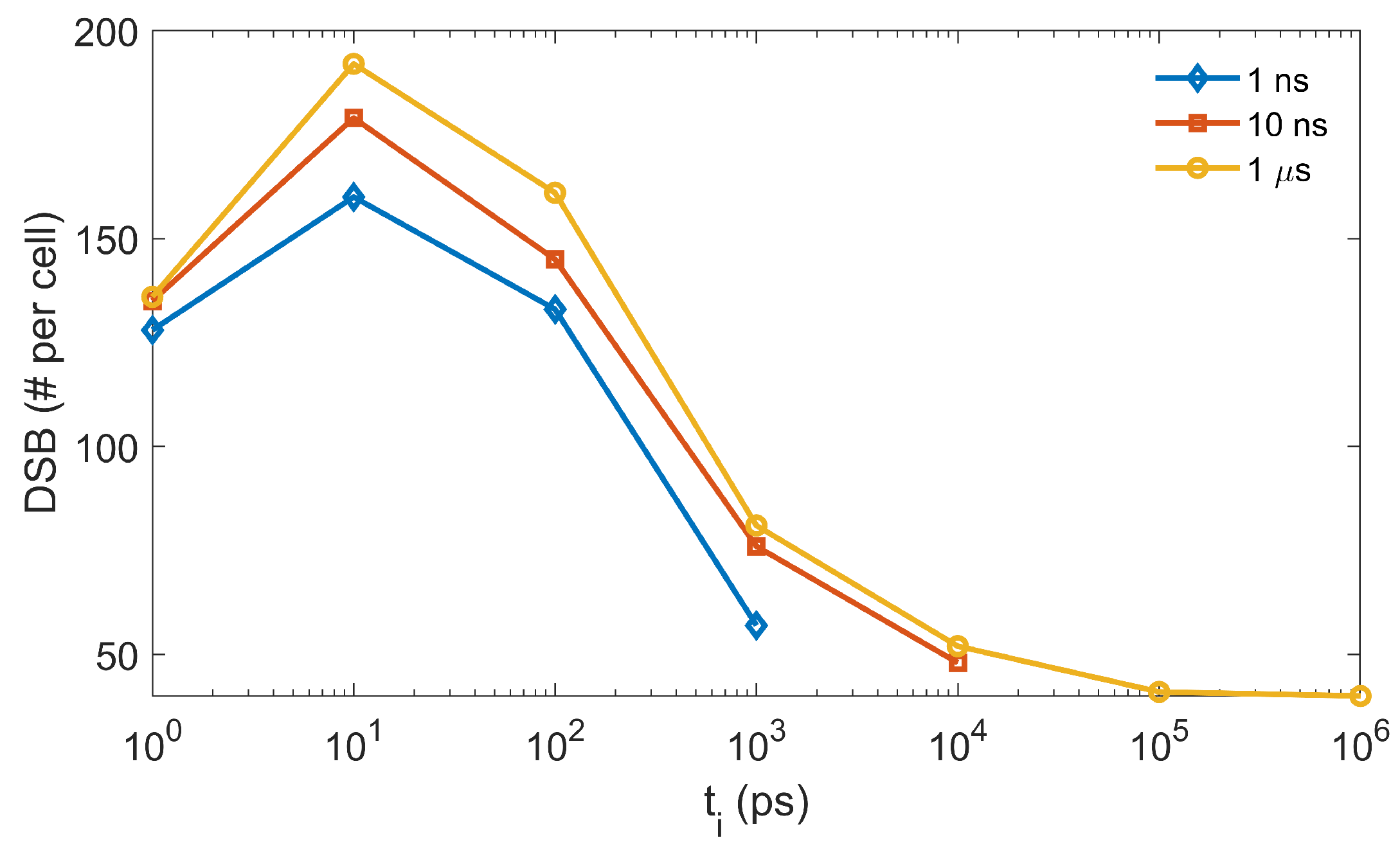
3.2. Validation of Concurrent Transport
3.3. Computational Efficiency
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GPU | Graphical Processing Unit |
| LET | Linear Energy Transfer |
| ROI | Region Of Interest |
| SDCS | Singly Differential Cross-Section |
| OER | Oxygen Enhancement Ratio |
| PSF | Phase Space File |
References
- Chatzipapas, K.P.; Papadimitroulas, P.; Emfietzoglou, D.; Kalospyros, S.A.; Hada, M.; Georgakilas, A.G.; Kagadis, G.C. Ionizing radiation and complex DNA damage: Quantifying the radiobiological damage using monte carlo simulations. Cancers 2020, 12, 799. [Google Scholar] [CrossRef] [Green Version]
- Paretzke, H.G. Radiation Track Structure Theory; Wiley: Hoboken, NJ, USA, 1987. [Google Scholar]
- Nikjoo, H.; O’Neill, P.; Goodhead, D.; Terrissol, M. Computational modeling of low-energy electron-induced DNA damage by early physical and chemical events. Int. J. Radiat. Biol. 1997, 71, 467–483. [Google Scholar] [CrossRef]
- Cucinotta, F.A.; Wilson, J.; Townsend, L.; Shinn, J.; Katz, R. Track structure model for damage to mammalian cell cultures during solar proton events. Int. J. Radiat. Appl. Instrum. Part D Nucl. Tracks Radiat. Meas. 1992, 20, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Michalik, V.; Běgusova, M. Target model of nucleosome particle for track structure calculations and DNA damage modeling. Int. J. Radiat. Biol. 1994, 66, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Uehara, S.; Emfietzoglou, D.; Cucinotta, F. Track-structure codes in radiation research. Radiat. Meas. 2006, 41, 1052–1074. [Google Scholar] [CrossRef]
- Friedland, W.; Dingfelder, M.; Kundrát, P.; Jacob, P. Track structures, DNA targets and radiation effects in the biophysical Monte Carlo simulation code PARTRAC. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Bernal, M.; Bordage, M.; Brown, J.; Davídková, M.; Delage, E.; El Bitar, Z.; Enger, S.; Francis, Z.; Guatelli, S.; Ivanchenko, V.; et al. Track structure modeling in liquid water: A review of the Geant4-DNA very low energy extension of the Geant4 Monte Carlo simulation toolkit. Phys. Med. 2015, 31, 861–874. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Tian, Z.; Qin, N.; Yan, C.; Lai, Y.; Hung, S.H.; Chi, Y.; Jia, X. A new open-source GPU-based microscopic Monte Carlo simulation tool for the calculations of DNA damages caused by ionizing radiation—Part I: Core algorithm and validation. Med. Phys. 2020, 47, 1958–1970. [Google Scholar] [CrossRef] [PubMed]
- Bordage, M.; Bordes, J.; Edel, S.; Terrissol, M.; Franceries, X.; Bardiès, M.; Lampe, N.; Incerti, S. Implementation of new physics models for low energy electrons in liquid water in Geant4-DNA. Phys. Med. 2016, 32, 1833–1840. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Kyriakou, I.; Garcia-Molina, R.; Abril, I.; Nikjoo, H. Inelastic cross-sections for low-energy electrons in liquid water: Exchange and correlation effects. Radiat. Res. 2013, 180, 499–513. [Google Scholar] [CrossRef]
- Kyriakou, I.; Incerti, S.; Francis, Z. Improvements in geant4 energy-loss model and the effect on low-energy electron transport in liquid water. Med. Phys. 2015, 42, 3870–3876. [Google Scholar] [CrossRef]
- Dingfelder, M. Updated model for dielectric response function of liquid water. Appl. Radiat. Isot. 2014, 83, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, T.; Yokoya, A.; Ukai, M.; Watanabe, R. Cross sections, stopping powers, and energy loss rates for rotational and phonon excitation processes in liquid water by electron impact. Radiat. Phys. Chem. 2015, 108, 13–17. [Google Scholar] [CrossRef]
- Shin, W.G.; Bordage, M.C.; Emfietzoglou, D.; Kyriakou, I.; Sakata, D.; Min, C.; Lee, S.B.; Guatelli, S.; Incerti, S. Development of a new Geant4-DNA electron elastic scattering model for liquid-phase water using the ELSEPA code. J. Appl. Phys. 2018, 124, 224901. [Google Scholar] [CrossRef] [Green Version]
- Pomplun, E. A new DNA target model for track structure calculations and its first application to I-125 Auger electrons. Int. J. Radiat. Biol. 1991, 59, 625–642. [Google Scholar] [CrossRef] [PubMed]
- Kreth, G.; Finsterle, J.; Von Hase, J.; Cremer, M.; Cremer, C. Radial arrangement of chromosome territories in human cell nuclei: A computer model approach based on gene density indicates a probabilistic global positioning code. Biophys. J. 2004, 86, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Schuemann, J.; McNamara, A.; Ramos-Méndez, J.; Perl, J.; Held, K.; Paganetti, H.; Incerti, S.; Faddegon, B. TOPAS-nBio: An extension to the TOPAS simulation toolkit for cellular and sub-cellular radiobiology. Radiat. Res. 2019, 191, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.H.; Conger, A.; Ebert, M.; Hornsey, S.; Scott, O. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Nair, C.K.; Parida, D.K.; Nomura, T. Radioprotectors in radiotherapy. J. Radiat. Res. 2001, 42, 21–37. [Google Scholar] [CrossRef]
- Boscolo, D.; Krämer, M.; Fuss, M.C.; Durante, M.; Scifoni, E. Impact of target oxygenation on the chemical track evolution of ion and electron radiation. Int. J. Mol. Sci. 2020, 21, 424. [Google Scholar] [CrossRef] [Green Version]
- Meylan, S.; Incerti, S.; Karamitros, M.; Tang, N.; Bueno, M.; Clairand, I.; Villagrasa, C. Simulation of early DNA damage after the irradiation of a fibroblast cell nucleus using Geant4-DNA. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Lampe, N.; Karamitros, M.; Breton, V.; Brown, J.M.; Kyriakou, I.; Sakata, D.; Sarramia, D.; Incerti, S. Mechanistic DNA damage simulations in Geant4-DNA part 1: A parameter study in a simplified geometry. Phys. Med. 2018, 48, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Sakata, D.; Incerti, S.; Bordage, M.; Lampe, N.; Okada, S.; Emfietzoglou, D.; Kyriakou, I.; Murakami, K.; Sasaki, T.; Tran, H.; et al. An implementation of discrete electron transport models for gold in the Geant4 simulation toolkit. J. Appl. Phys. 2016, 120, 244901. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Ziegenhein, P.; Jiang, S.B. GPU-based high-performance computing for radiation therapy. Phys. Med. Biol. 2014, 59, R151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Després, P.; Jia, X. A review of GPU-based medical image reconstruction. Phys. Med. 2017, 42, 76–92. [Google Scholar] [CrossRef]
- Tian, Z.; Jiang, S.B.; Jia, X. Accelerated Monte Carlo simulation on the chemical stage in water radiolysis using GPU. Phys. Med. Biol. 2017, 62, 3081. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Jia, X.; Chi, Y. Modeling the effect of oxygen on the chemical stage of water radiolysis using GPU-based microscopic Monte Carlo simulations, with an application in FLASH radiotherapy. Phys. Med. Biol. 2021, 66, 025004. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Tsai, M.Y.; Tian, Z.; Qin, N.; Yan, C.; Hung, S.H.; Chi, Y.; Jia, X. A new open-source GPU-based microscopic Monte Carlo simulation tool for the calculations of DNA damages caused by ionizing radiation—Part II: Sensitivity and uncertainty analysis. Med. Phys. 2020, 47, 1971–1982. [Google Scholar] [CrossRef]
- Paganetti, H.; Niemierko, A.; Ancukiewicz, M.; Gerweck, L.E.; Goitein, M.; Loeffler, J.S.; Suit, H.D. Relative biological effectiveness (RBE) values for proton beam therapy. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 407–421. [Google Scholar] [CrossRef]
- Vitti, E.T.; Parsons, J.L. The radiobiological effects of proton beam therapy: Impact on DNA damage and repair. Cancers 2019, 11, 946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, K.G.; Kanai, Y. Electronic excitation dynamics in liquid water under proton irradiation. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, M.E.; Kim, Y.K.; Madison, D.H.; Gay, T.J. Electron production in proton collisions with atoms and molecules: Energy distributions. Rev. Mod. Phys. 1992, 64, 441. [Google Scholar] [CrossRef] [Green Version]
- Plante, I.; Cucinotta, F.A. Ionization and excitation cross-sections for the interaction of HZE particles in liquid water and application to Monte Carlo simulation of radiation tracks. New J. Phys. 2008, 10, 125020. [Google Scholar] [CrossRef] [Green Version]
- Dingfelder, M.; Inokuti, M.; Paretzke, H.G. Inelastic-collision cross-sections of liquid water for interactions of energetic protons. Radiat. Phys. Chem. 2000, 59, 255–275. [Google Scholar] [CrossRef]
- Booth, W.; Grant, I. The energy loss of oxygen and chlorine ions in solids. Nucl. Phys. 1965, 63, 481–495. [Google Scholar] [CrossRef]
- Dingfelder, M.; Jorjishvili, I.; Gersh, J.; Toburen, L. Heavy ion track structure simulations in liquid water at relativistic energies. Radiat. Prot. Dosim. 2006, 122, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M. Cross sections for production of secondary electrons by charged particles. Radiat. Prot. Dosim. 1990, 31, 17–22. [Google Scholar] [CrossRef]
- Kutcher, G.; Green, A. A model for energy deposition in liquid water. Radiat. Res. 1976, 67, 408–425. [Google Scholar] [CrossRef]
- Cobut, V.; Frongillo, Y.; Patau, J.; Goulet, T.; Fraser, M.; Jay-Gerin, J. Monte Carlo simulation of fast electron and proton tracks in liquid water-I. Physical and physicochemical aspects. Radiat. Phys. Chem. 1998, 51, 229–244. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅ OH/⋅ O- in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; McNamara, A.L.; Ramos-Mendez, J.; McMahon, S.J.; Henthorn, N.T.; Faddegon, B.; Held, K.D.; Perl, J.; Li, J.; Paganetti, H.; et al. A parameter sensitivity study for simulating DNA damage after proton irradiation using TOPAS-nBio. Phys. Med. Biol. 2020, 65, 085015. [Google Scholar] [CrossRef]
- Incerti, S.; Baldacchino, G.; Bernal, M.; Capra, R.; Champion, C.; Francis, Z.; Guèye, P.; Mantero, A.; Mascialino, B.; Moretto, P.; et al. The geant4-dna project. Int. J. Model. Simul. Sci. Comput. 2010, 1, 157–178. [Google Scholar] [CrossRef]
- Wang, H.; Vassiliev, O.N. Radial dose distributions from protons of therapeutic energies calculated with Geant4-DNA. Phys. Med. Biol. 2014, 59, 3657. [Google Scholar] [CrossRef] [Green Version]
- Nikjoo, H.; O’Neill, P.; Wilson, W.; Goodhead, D. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Nikjoo, H.; Emfietzoglou, D.; Liamsuwan, T.; Taleei, R.; Liljequist, D.; Uehara, S. Radiation track, DNA damage and response—A review. Rep. Prog. Phys. 2016, 79, 116601. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Emfietzoglou, D.; Watanabe, R.; Uehara, S. Can Monte Carlo track structure codes reveal reaction mechanism in DNA damage and improve radiation therapy? Radiat. Phys. Chem. 2008, 77, 1270–1279. [Google Scholar] [CrossRef]
- Lemelin, V.; Bass, A.; Cloutier, P.; Sanche, L. Low energy (1–19 eV) electron scattering from condensed thymidine (dT) I: Absolute vibrational excitation cross-sections. Phys. Chem. Chem. Phys. 2019, 21, 23808–23817. [Google Scholar] [CrossRef] [PubMed]
- Emfietzoglou, D.; Papamichael, G.; Nikjoo, H. Monte Carlo electron track structure calculations in liquid water using a new model dielectric response function. Radiat. Res. 2017, 188, 355–368. [Google Scholar] [CrossRef]
- Ramos-Méndez, J.; Shin, W.g.; Karamitros, M.; Domínguez-Kondo, J.; Tran, N.H.; Incerti, S.; Villagrasa, C.; Perrot, Y.; Štěpán, V.; Okada, S.; et al. Independent reaction times method in Geant4-DNA: Implementation and performance. Med. Phys. 2020, 47, 5919–5930. [Google Scholar] [CrossRef] [PubMed]
- Liamsuwan, T.; Emfietzoglou, D.; Uehara, S.; Nikjoo, H. Microdosimetry of low-energy electrons. Int. J. Radiat. Biol. 2012, 88, 899–907. [Google Scholar] [CrossRef]
- Shin, W.G.; Ramos-Mendez, J.; Faddegon, B.; Tran, H.; Villagrasa, C.; Perrot, Y.; Okada, S.; Karamitros, M.; Emfietzoglou, D.; Kyriakou, I.; et al. Evaluation of the influence of physical and chemical parameters on water radiolysis simulations under MeV electron irradiation using Geant4-DNA. J. Appl. Phys. 2019, 126, 114301. [Google Scholar] [CrossRef]
- Nikjoo, H.; Uehara, S.; Emfietzoglou, D.; Brahme, A. Heavy charged particles in radiation biology and biophysics. New J. Phys. 2008, 10, 075006. [Google Scholar] [CrossRef]
- Kyriakou, I.; Šefl, M.; Nourry, V.; Incerti, S. The impact of new Geant4-DNA cross-section models on electron track structure simulations in liquid water. J. Appl. Phys. 2016, 119, 194902. [Google Scholar] [CrossRef]
- Incerti, S.; Kyriakou, I.; Bernal, M.; Bordage, M.C.; Francis, Z.; Guatelli, S.; Ivanchenko, V.; Karamitros, M.; Lampe, N.; Lee, S.B.; et al. Geant4-DNA example applications for track structure simulations in liquid water: A report from the Geant4-DNA Project. Med. Phys. 2018, 45, e722–e739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuya, Y.; Kai, T.; Yoshii, Y.; Yachi, Y.; Naijo, S.; Date, H.; Sato, T. Modeling of yield estimation for DNA strand breaks based on Monte Carlo simulations of electron track structure in liquid water. J. Appl. Phys. 2019, 126, 124701. [Google Scholar] [CrossRef]
- Friedland, W.; Schmitt, E.; Kundrát, P.; Dingfelder, M.; Baiocco, G.; Barbieri, S.; Ottolenghi, A. Comprehensive track-structure based evaluation of DNA damage by light ions from radiotherapy-relevant energies down to stopping. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Aydogan, B.; Bolch, W.E.; Swarts, S.G.; Turner, J.E.; Marshall, D.T. Monte Carlo simulations of site-specific radical attack to DNA bases. Radiat. Res. 2008, 169, 223–231. [Google Scholar] [CrossRef]
- Verlackt, C.; Neyts, E.; Jacob, T.; Fantauzzi, D.; Golkaram, M.; Shin, Y.; Van Duin, A.; Bogaerts, A. Atomic-scale insight into the interactions between hydroxyl radicals and DNA in solution using the ReaxFF reactive force field. New J. Phys. 2015, 17, 103005. [Google Scholar] [CrossRef]
- Schuemann, J.; McNamara, A.; Warmenhoven, J.; Henthorn, N.; Kirkby, K.J.; Merchant, M.J.; Ingram, S.; Paganetti, H.; Held, K.D.; Ramos-Mendez, J.; et al. A new standard DNA damage (SDD) data format. Radiat. Res. 2019, 191, 76–92. [Google Scholar] [CrossRef]
- Kalantzis, G.; Emfietzoglou, D.; Hadjidoukas, P. A unified spatio-temporal parallelization framework for accelerated Monte Carlo radiobiological modeling of electron tracks and subsequent radiation chemistry. Comput. Phys. Commun. 2012, 183, 1683–1695. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Inner Orbitals | External Orbitals | |||
|---|---|---|---|---|---|
| 1.25 | 1.25 | 1.02 | 1.02 | 1.02 | |
| 0.5 | 0.5 | 82 | 82 | 82 | |
| 1 | 1 | 0.45 | 0.45 | 0.45 | |
| 1 | 1 | −0.80 | −0.80 | −0.80 | |
| 3 | 3 | 0.38 | 0.38 | 0.38 | |
| 1.1 | 1.1 | 1.07 | 1.07 | 1.07 | |
| 1.3 | 1.3 | 14.6 | 14.6 | 14.6 | |
| 1 | 1 | 0.6 | 0.6 | 0.6 | |
| 0 | 0 | 0.04 | 0.04 | 0.04 | |
| 0.66 | 0.66 | 0.64 | 0.64 | 0.64 | |
| 2 | 2 | 2 | 2 | 2 | |
| 539.7 | 32.2 | 18.55 | 14.73 | 12.61 | |
| j | Plasma Mode | ||
|---|---|---|---|
| 0.0187 | 0.0157 | 0.7843 | |
| 3 (eV) | 1 (eV) | 0.6 (eV) | |
| 8.4 | 10.1 | 21.3 |
| Radicals | A | G | C | T | DNA Base | DNA Sugar-Phosphate Group |
|---|---|---|---|---|---|---|
| 6.1 | 9.2 | 6.4 | 6.1 | 6.95 | 1.9 | |
| 9 | 14 | 18 | 13 | 13.5 | * |
| Energy (MeV) | from gMicroMC | from Nikjoo’s Work |
|---|---|---|
| 0.9 | 20.1 | 18.2 |
| 0.5 | 25.1 | 23.9 |
| Energy (MeV) | Number of Primary Protons | ||
|---|---|---|---|
| 1 | 10 | 100 | |
| 1 | 1.9 | 3.1 | 9.3 |
| 10 | 3.9 | 9.8 | 40.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, Y.; Jia, X.; Chi, Y. Recent Developments on gMicroMC: Transport Simulations of Proton and Heavy Ions and Concurrent Transport of Radicals and DNA. Int. J. Mol. Sci. 2021, 22, 6615. https://doi.org/10.3390/ijms22126615
Lai Y, Jia X, Chi Y. Recent Developments on gMicroMC: Transport Simulations of Proton and Heavy Ions and Concurrent Transport of Radicals and DNA. International Journal of Molecular Sciences. 2021; 22(12):6615. https://doi.org/10.3390/ijms22126615
Chicago/Turabian StyleLai, Youfang, Xun Jia, and Yujie Chi. 2021. "Recent Developments on gMicroMC: Transport Simulations of Proton and Heavy Ions and Concurrent Transport of Radicals and DNA" International Journal of Molecular Sciences 22, no. 12: 6615. https://doi.org/10.3390/ijms22126615
APA StyleLai, Y., Jia, X., & Chi, Y. (2021). Recent Developments on gMicroMC: Transport Simulations of Proton and Heavy Ions and Concurrent Transport of Radicals and DNA. International Journal of Molecular Sciences, 22(12), 6615. https://doi.org/10.3390/ijms22126615