
Combined Gemcitabine and Immune-Checkpoint Inhibition Conquers Anti-PD-L1 Resistance in Low-Immunogenic Mismatch Repair-Deficient Tumors

 , , , ,
, , , ,
Abstract
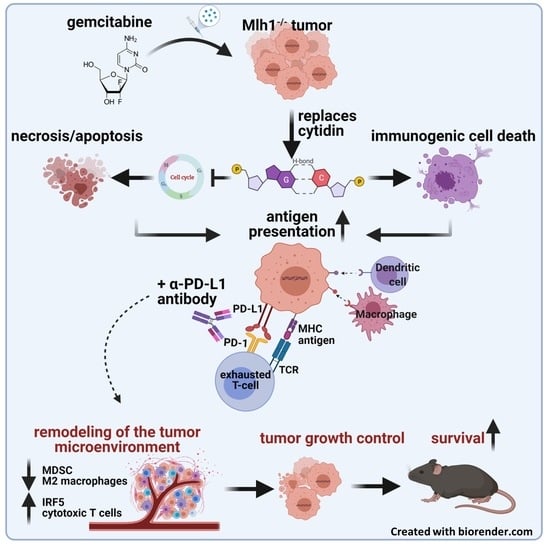
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. In Vitro Analysis
2.2. Combination of α-PD-L1 and CTX Prolongs the Survival of Mlh1−/− Mice
2.3. Peripheral Immune Activation by Combinational Therapy
2.4. Spleens and Residual Tumors Change Their Immunological Profile
2.5. Treatment with α-PD-L1 Induced Molecular Changes in cMS of dMMR-Target Genes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Colony Formation Assay
4.3. Co-Culture Experiments
4.4. Mlh1−/− Mouse Model and In Vivo Treatment Protocol
4.4.1. Institutional Review Board Statement
4.4.2. Experimental Protocol
4.5. PET/CT Imaging
4.6. Immune Phenotyping
4.7. Procartaplex Cytokine Assay
4.8. Fragment Length Analysis of cMS Target Genes
4.9. Immunofluorescence
4.10. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ning, Y.; Suzman, D.; Maher, V.E.; Zhang, L.; Tang, S.; Ricks, T.; Palmby, T.; Fu, W.; Liu, Q.; Goldberg, K.B.; et al. FDA Approval Summary: Atezolizumab for the Treatment of Patients with Progressive Advanced Urothelial Carcinoma after Platinum-Containing Chemotherapy. Oncologist 2017, 22, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Hoffman-Censits, J.; Zheng, H.; Kaiser, C.; Tayama, D.; Bellmunt, J. Atezolizumab in Platinum-treated Locally Advanced or Metastatic Urothelial Carcinoma: Clinical Experience from an Expanded Access Study in the United States. Eur. Urol. 2018, 73, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.; Young, R.J.; Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin. Cancer Biol. 2018, 52, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Álvaro, E.; Cano, J.M.; García, J.L.; Brandáriz, L.; Olmedillas-López, S.; Arriba, M.; Rueda, D.; Rodríguez, Y.; Cañete, Á.; Arribas, J.; et al. Clinical and molecular comparative study of colorectal cancer based on age-of-onset and tumor location: Two main criteria for subclassifying colorectal cancer. Int. J. Mol. Sci. 2019, 20, 968. [Google Scholar] [CrossRef] [Green Version]
- Evrard, C.; Tachon, G.; Randrian, V.; Karayan-Tapon, L.; Tougeron, D. Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers 2019, 11, 1567. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin. Genet. 2017, 91, 507–519. [Google Scholar] [CrossRef]
- Tabori, U.; Hansford, J.R.; Achatz, M.I.; Kratz, C.P.; Plon, S.E.; Frebourg, T.; Brugieres, L. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin. Cancer Res. 2017, 23, e32–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakry, D.; Aronson, M.; Durno, C.; Rimawi, H.; Farah, R.; Alharbi, Q.K.; Alharbi, M.; Shamvil, A.; Ben-Shachar, S.; Mistry, M.; et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: Report from the constitutional mismatch repair deficiency consortium. Eur. J. Cancer 2014, 50, 987–996. [Google Scholar] [CrossRef]
- Aronson, M.; Colas, C.; Shuen, A.; Hampel, H.; Foulkes, W.D.; Baris Feldman, H.; Goldberg, Y.; Muleris, M.; Wolfe Schneider, K.; McGee, R.B.; et al. Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): Recommendations from the international consensus working group. J. Med. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Sahin, I.H. Defining the current role of immune checkpoint inhibitors in the treatment of mismatch repair-deficient/microsatellite stability-high colorectal cancer and shedding light on future approaches. Expert Rev. Gastroenterol. Hepatol. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Trullas, A.; Delgado, J.; Genazzani, A.; Mueller-Berghaus, J.; Migali, C.; Müller-Egert, S.; Zander, H.; Enzmann, H.; Pignatti, F. The EMA assessment of pembrolizumab as monotherapy for the first-line treatment of adult patients with metastatic microsatellite instability-high or mismatch repair deficient colorectal cancer. ESMO Open 2021, 6, 100145. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, J.; André, T.; El Hajbi, F.; Barbier, E.; Toullec, C.; Kim, S.; Bouche, O.; Di Fiore, F.; Chauvenet, M.; Perrier, H.; et al. Avelumab versus standard second line treatment chemotherapy in metastatic colorectal cancer patients with microsatellite instability: The SAMCO-PRODIGE 54 randomised phase II trial. Dig. Liver Dis. 2020, 53, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Chen, Y.-P.; Li, Y.-Q.; Liu, N.; Ma, J. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol. Cancer 2021, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Ager, A.; Arends, M.J.; Frayling, I.M. Lynch syndrome—Cancer pathways, heterogeneity and immune escape. J. Pathol. 2018, 246, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Juhling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Parente, P.; Parcesepe, P.; Covelli, C.; Olivieri, N.; Remo, A.; Pancione, M.; Latiano, T.P.; Graziano, P.; Maiello, E.; Giordano, G. Crosstalk between the tumor microenvironment and immune system in pancreatic ductal adenocarcinoma: Potential targets for new therapeutic approaches. Gastroenterol. Res. Pract. 2018, 2018. [Google Scholar] [CrossRef]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef]
- Maletzki, C.; Wiegele, L.; Nassar, I.; Stenzel, J.; Junghanss, C. Chemo-immunotherapy improves long-term survival in a preclinical model of MMR-D-related cancer. J. Immunother. Cancer 2019, 7, 1–4. [Google Scholar] [CrossRef]
- Salewski, I.; Gladbach, Y.S.; Kuntoff, S.; Irmscher, N.; Hahn, O.; Junghanss, C.; Maletzki, C. In vivo vaccination with cell line-derived whole tumor lysates: Neoantigen quality, not quantity matters. J. Transl. Med. 2020, 18, 1–5. [Google Scholar] [CrossRef]
- Gladbach, Y.S.; Wiegele, L.; Hamed, M.; Merkenschläger, A.M.; Fuellen, G.; Junghanss, C.; Maletzki, C. Unraveling the Heterogeneous Mutational Signature of Spontaneously Developing Tumors in MLH1−/− Mice. Cancers 2019, 11, 1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, J.P.H.; Todd Reilly, R.T.; Emens, L.A.; Ercolini, A.M.; Lei, R.Y.; Weintraub, D.; Okoye, F.I.; Jaffee, E.M. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001, 61, 3689–3697. [Google Scholar]
- Sukkurwala, A.Q.; Adjemian, S.; Senovilla, L.; Michaud, M.; Spaggiari, S.; Vacchelli, E.; Baracco, E.E.; Galluzzi, L.; Zitvogel, L.; Kepp, O.; et al. Screening of novel immunogenic cell death inducers within the NCI mechanistic diversity set. Oncoimmunology 2014, 3, e28473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hato, S.V.; Khong, A.; De Vries, I.J.M.; Lesterhuis, W.J. Molecular pathways: The immunogenic effects of platinum-based chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebremeskel, S.; Johnston, B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: Impact on clinical studies and considerations for combined therapies. Oncotarget 2015, 6, 41600–41619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G. The Mechanisms Leading to Distinct Responses to PD-1/PD-L1 Blockades in Colorectal Cancers With Different MSI Statuses. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Wang, T.; Lee, L.H.; Vyas, M.; Zhang, L.; Ganesh, K.; Firat, C.; Segal, N.H.; Desai, A.; Hechtman, J.F.; Ntiamoah, P.; et al. Colorectal carcinoma with double somatic mismatch repair gene inactivation: Clinical and pathological characteristics and response to immune checkpoint blockade. Mod. Pathol. 2019, 32, 1551–1562. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/ mismatch repair–deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Brahmer, J.R.; Juergens, R.A.; Borghaei, H.; Gettinger, S.; Chow, L.Q.; Gerber, D.E.; Laurie, S.A.; Goldman, J.W.; et al. Nivolumab in combination with platinum-based doublet chemotherapy for first-line treatment of advanced non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Kanda, S.; Goto, K.; Shiraishi, H.; Kubo, E.; Tanaka, A.; Utsumi, H.; Sunami, K.; Kitazono, S.; Mizugaki, H.; Horinouchi, H.; et al. Safety and efficacy of nivolumab and standard chemotherapy drug combination in patients with advanced non-small-cell lung cancer: A four arms phase Ib study. Ann. Oncol. 2016, 27, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Aoe, K.; Kozuki, T.; Oze, I.; Kato, K.; Kishimoto, T.; Hotta, K. A Phase II Trial of First-Line Combination Chemotherapy With Cisplatin, Pemetrexed, and Nivolumab for Unresectable Malignant Pleural Mesothelioma: A Study Protocol. Clin. Lung Cancer 2018, 19, e705–e707. [Google Scholar] [CrossRef]
- Dudnik, E.; Moskovitz, M.; Rottenberg, Y.; Lobachov, A.; Mandelboim, R.; Shochat, T.; Urban, D.; Wollner, M.; Nechushtan, H.; Rotem, O.; et al. Pembrolizumab as a monotherapy or in combination with platinum-based chemotherapy in advanced non-small cell lung cancer with PD-L1 tumor proportion score (TPS) ≥50%: Real-world data. Oncoimmunology 2021, 10. [Google Scholar] [CrossRef]
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F.; et al. Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res. 2020, 80, 3101–3115. [Google Scholar] [CrossRef] [Green Version]
- Baba, Y.; Nomoto, D.; Okadome, K.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Baba, H. Tumor immune microenvironment and immune checkpoint inhibitors in esophageal squamous cell carcinoma. Cancer Sci. 2020, 111, 3132–3141. [Google Scholar] [CrossRef] [PubMed]
- Seledtsov, V.I.; Goncharov, A.G.; Seledtsova, G.V. Clinically feasible approaches to potentiating cancer cell-based immunotherapies. Hum. Vaccines Immunother. 2015, 11, 851–869. [Google Scholar] [CrossRef] [Green Version]
- Martens, A.; Wistuba-Hamprecht, K.; Geukes Foppen, M.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wei, G.; Cheng, W.A.; Dong, Z.; Sun, H.; Lee, V.Y.; Cha, S.-C.; Smith, D.L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1181–1195. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Lee, B.-H.; Kim, S.-E.; Kwon, B.-E.; Jeong, H.; Choi, J.R.; Kim, M.J.; Park, Y.; Kim, B.S.; Kim, D.H.; et al. A Novel Anti-PD-L1 Antibody Exhibits Antitumor Effects on Multiple Myeloma in Murine Models via Antibody-Dependent Cellular Cytotoxicity. Biomol. Ther. 2020, 29, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Arasanz, H.; Chocarro, L.; Bocanegra, A.; Zuazo, M.; Fernandez-Hinojal, G.; Blanco, E.; Vera, R.; Escors, D.; Kochan, G. Systemic blood immune cell populations as biomarkers for the outcome of immune checkpoint inhibitor therapies. Int. J. Mol. Sci. 2020, 21, 2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1014–1030. [Google Scholar] [CrossRef] [Green Version]
- Weiss, M.; Blazek, K.; Byrne, A.J.; Perocheau, D.P.; Udalova, I.A. IRF5 Is a Specific Marker of Inflammatory Macrophages In Vivo. Mediat. Inflamm. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kwong, T.T.; Wong, C.H.; Zhou, J.Y.; Cheng, A.S.L.; Sung, J.J.Y.; Chan, A.W.H.; Chan, S.L. Chemotherapy-induced recruitment of myeloid-derived suppressor cells abrogates efficacy of immune checkpoint blockade. JHEP Rep. 2021, 3, 100224. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Sun, J.; Kapoor, V.; Jassar, A.S.; Albelda, S.M. Gemcitabine has significant immunomodulatory activity in murine tumor models independent of its cytotoxic effects. Cancer Biol. Ther. 2007, 6, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.; Al-Ghawi, H.; Helft, P.; House, M.G.; Spittler, A.J.; Wu, H.H.; Shahda, S. Two Months of Therapy: A Case of Pathologic Complete Response to Chemoimmunotherapy in a Patient With Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2018, 17, e229–e232. [Google Scholar] [CrossRef] [Green Version]
- Lizardo, D.Y.; Kuang, C.; Hao, S.; Yu, J.; Huang, Y.; Zhang, L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188447. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Luo, W.; Liu, J.F.; Gulhan, D.C.; Krasner, C.; Ishizuka, J.J.; Gockley, A.A.; Buss, M.; Growdon, W.B.; Crowe, H.; et al. Phase II study of avelumab in patients with mismatch repair deficient and mismatch repair proficient recurrent/persistent endometrial cancer. J. Clin. Oncol. 2019, 37, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Beyrich, F.; Hühns, M.; Klar, E.; Linnebacher, M. The mutational profile and infiltration pattern of murine MLH1−/− tumors: Concurrences, disparities and cell line establishment for functional analysis. Oncotarget 2016, 7, 53583–53598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Gladbach, Y.S.; Hamed, M.; Fuellen, G.; Semmler, M.-L.; Stenzel, J.; Linnebacher, M. Cellular vaccination of MLH1−/−mice–an immunotherapeutic proof of concept study. Oncoimmunology 2018, 7, e1408748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, S.; Lindner, T.; Polei, S.; Stenzel, J.; Borufka, L.; Achilles, S.; Hartmann, E.; Lange, F.; Maletzki, C.; Linnebacher, M.; et al. Application of in vivo imaging techniques to monitor therapeutic efficiency of PLX4720 in an experimental model of microsatellite instable colorectal cancer. Oncotarget 2017, 8, 69756–69767. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salewski, I.; Henne, J.; Engster, L.; Schneider, B.; Lemcke, H.; Skorska, A.; Berlin, P.; Henze, L.; Junghanss, C.; Maletzki, C. Combined Gemcitabine and Immune-Checkpoint Inhibition Conquers Anti-PD-L1 Resistance in Low-Immunogenic Mismatch Repair-Deficient Tumors. Int. J. Mol. Sci. 2021, 22, 5990. https://doi.org/10.3390/ijms22115990
Salewski I, Henne J, Engster L, Schneider B, Lemcke H, Skorska A, Berlin P, Henze L, Junghanss C, Maletzki C. Combined Gemcitabine and Immune-Checkpoint Inhibition Conquers Anti-PD-L1 Resistance in Low-Immunogenic Mismatch Repair-Deficient Tumors. International Journal of Molecular Sciences. 2021; 22(11):5990. https://doi.org/10.3390/ijms22115990
Chicago/Turabian StyleSalewski, Inken, Julia Henne, Leonie Engster, Bjoern Schneider, Heiko Lemcke, Anna Skorska, Peggy Berlin, Larissa Henze, Christian Junghanss, and Claudia Maletzki. 2021. "Combined Gemcitabine and Immune-Checkpoint Inhibition Conquers Anti-PD-L1 Resistance in Low-Immunogenic Mismatch Repair-Deficient Tumors" International Journal of Molecular Sciences 22, no. 11: 5990. https://doi.org/10.3390/ijms22115990
APA StyleSalewski, I., Henne, J., Engster, L., Schneider, B., Lemcke, H., Skorska, A., Berlin, P., Henze, L., Junghanss, C., & Maletzki, C. (2021). Combined Gemcitabine and Immune-Checkpoint Inhibition Conquers Anti-PD-L1 Resistance in Low-Immunogenic Mismatch Repair-Deficient Tumors. International Journal of Molecular Sciences, 22(11), 5990. https://doi.org/10.3390/ijms22115990