
Nrf2 Activation Attenuates Acrylamide-Induced Neuropathy in Mice †

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Changes in Body Weight
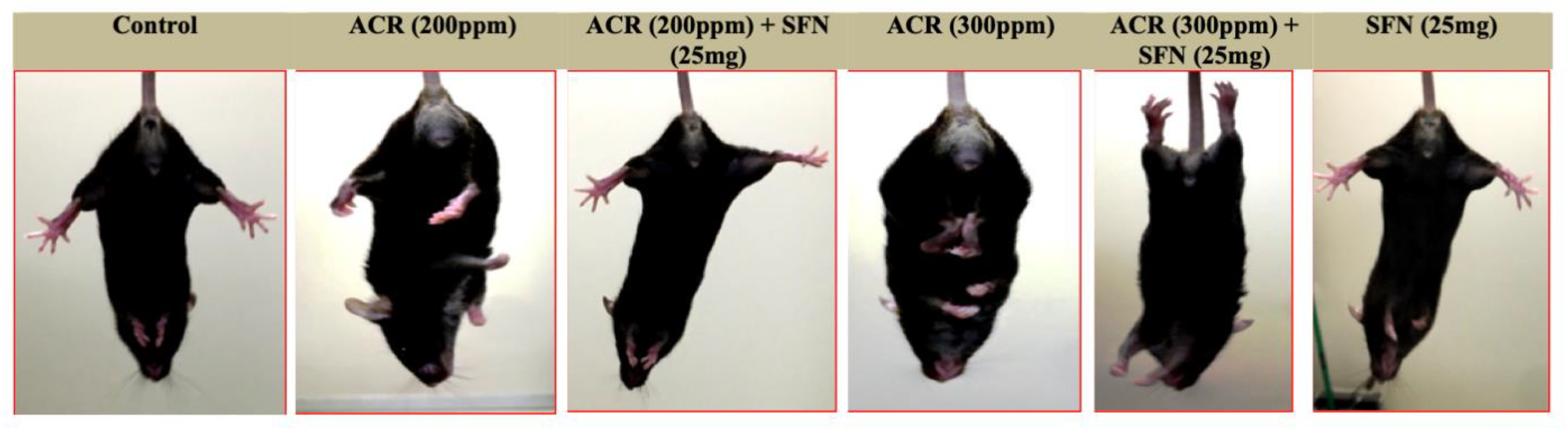
2.2. Changes in Function
Landing Foot Spread and Hindlimb Clasping Effect Observation
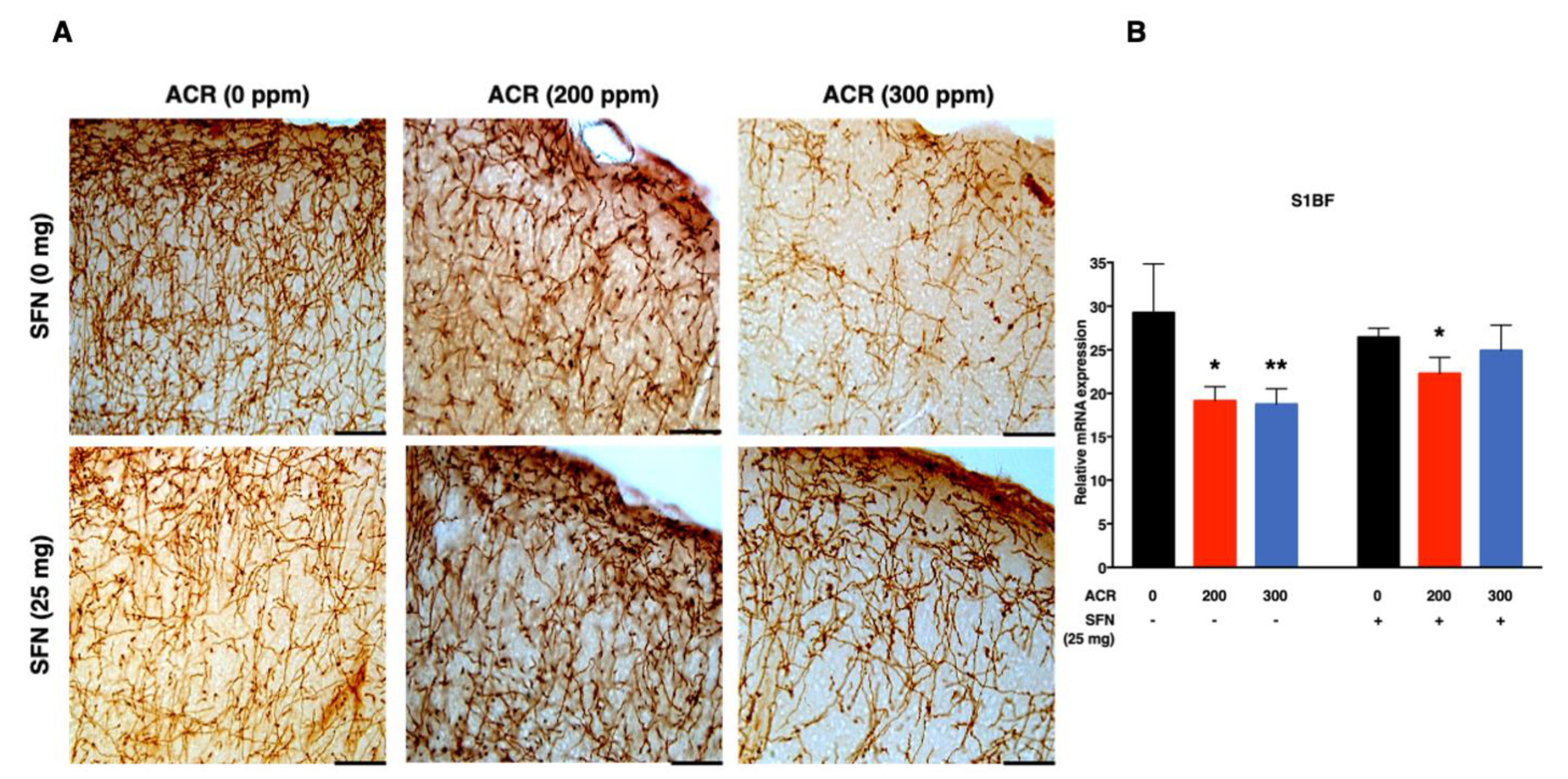
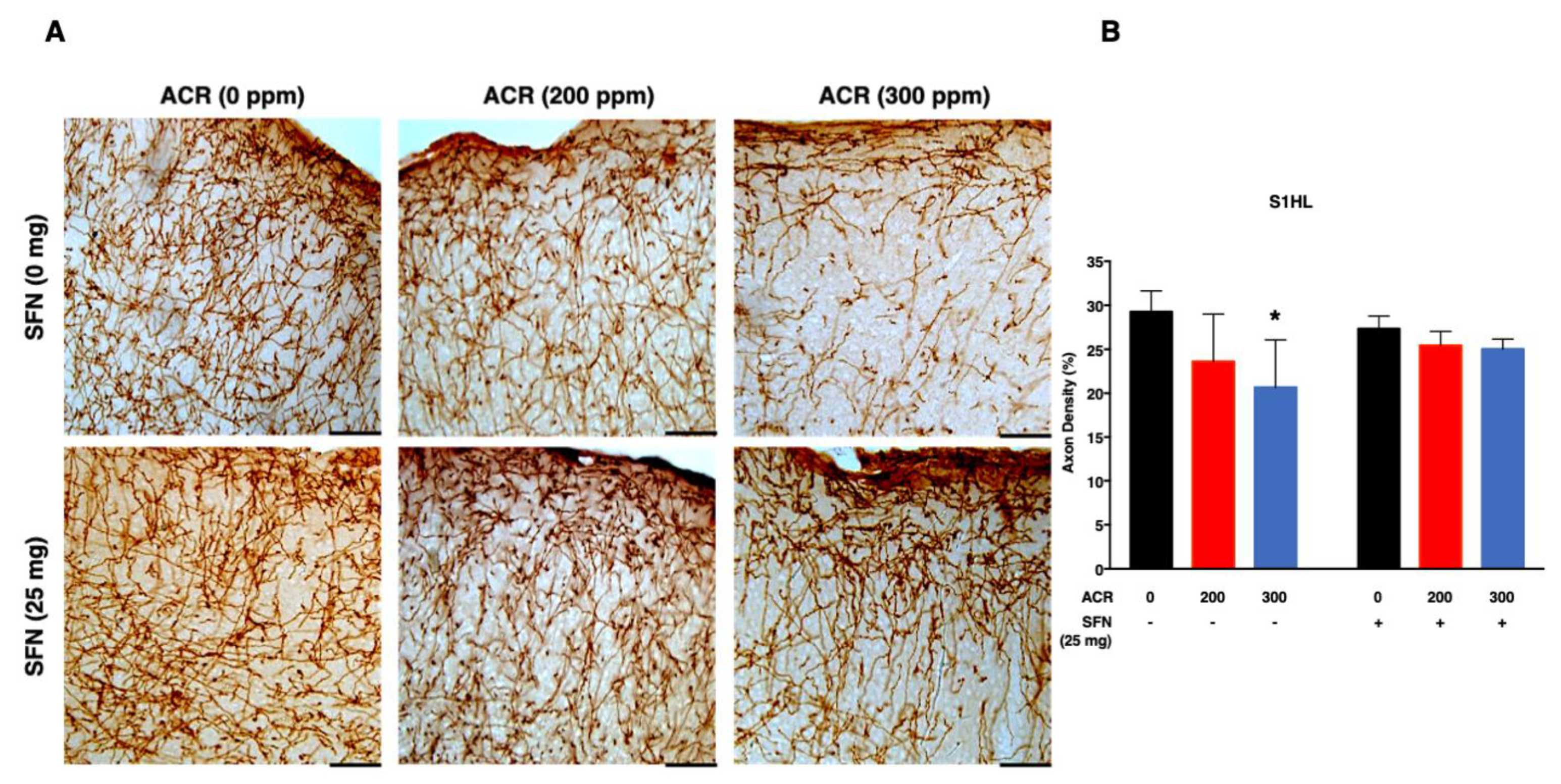
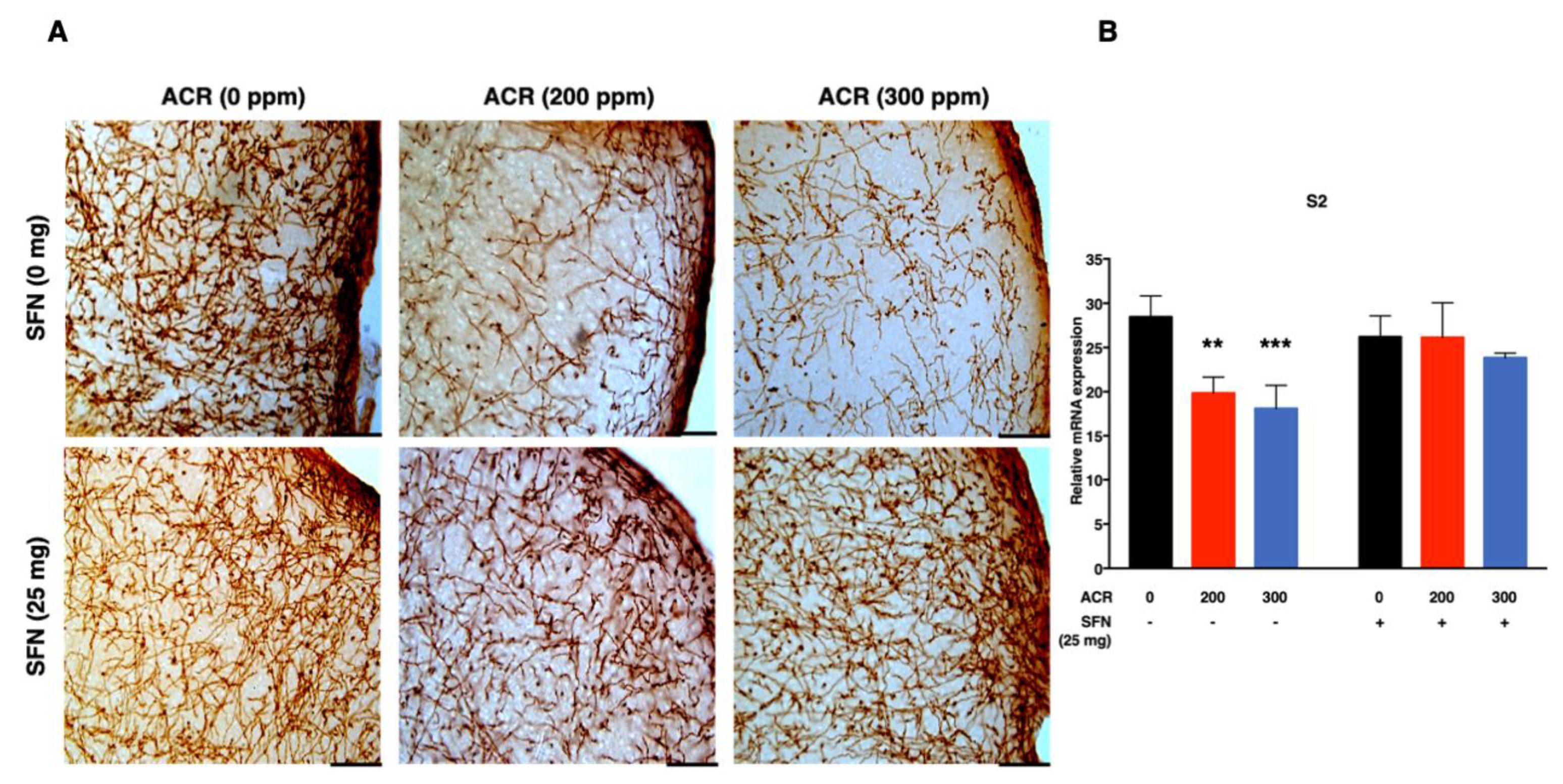
2.3. Changes in Monoaminergic Axons
Noradrenaline Transporter (NAT)-Immunoreactive (Noradrenergic) Axons
2.4. Changes in mRNA Expression
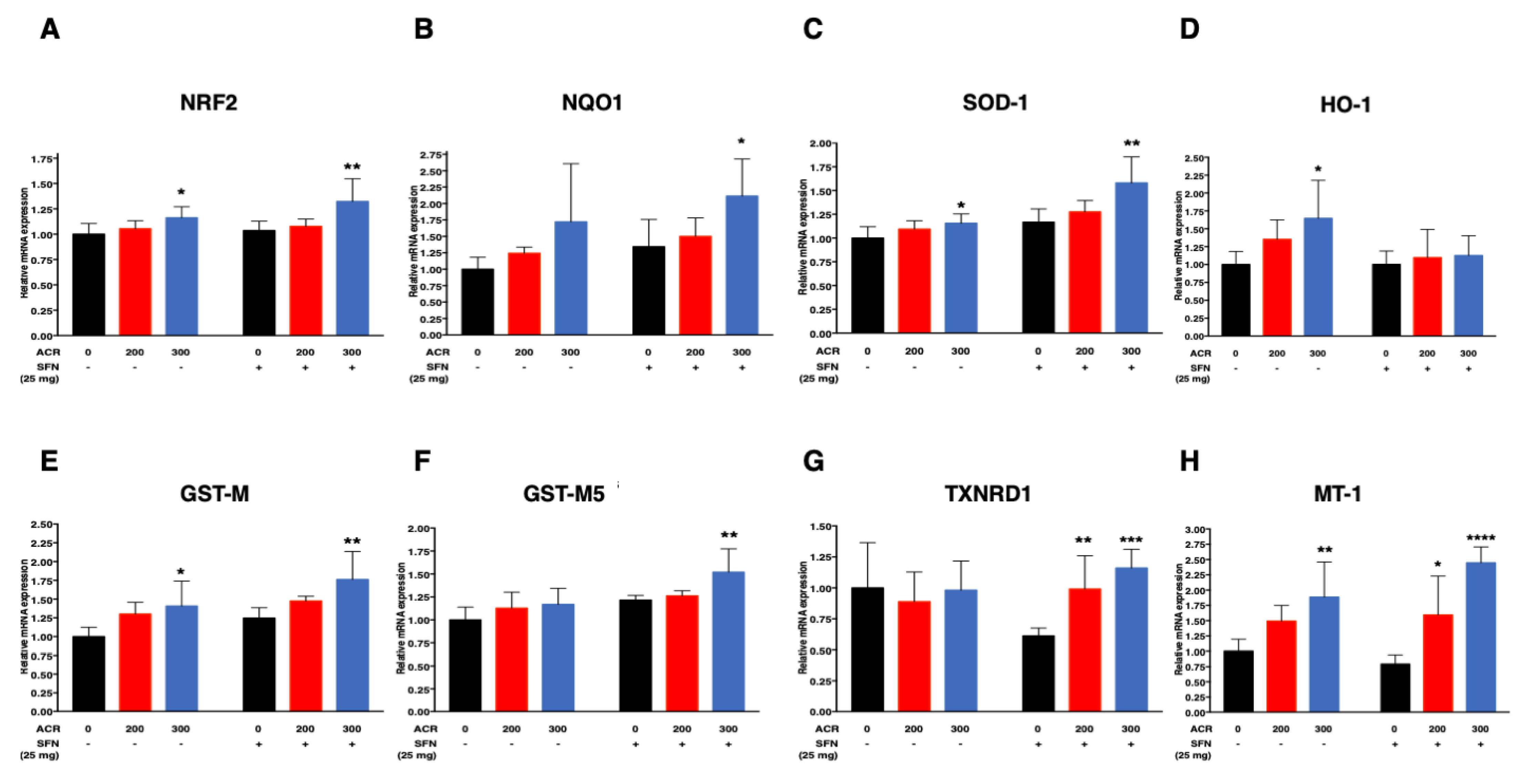
2.4.1. Nrf2-Antioxidant Genes
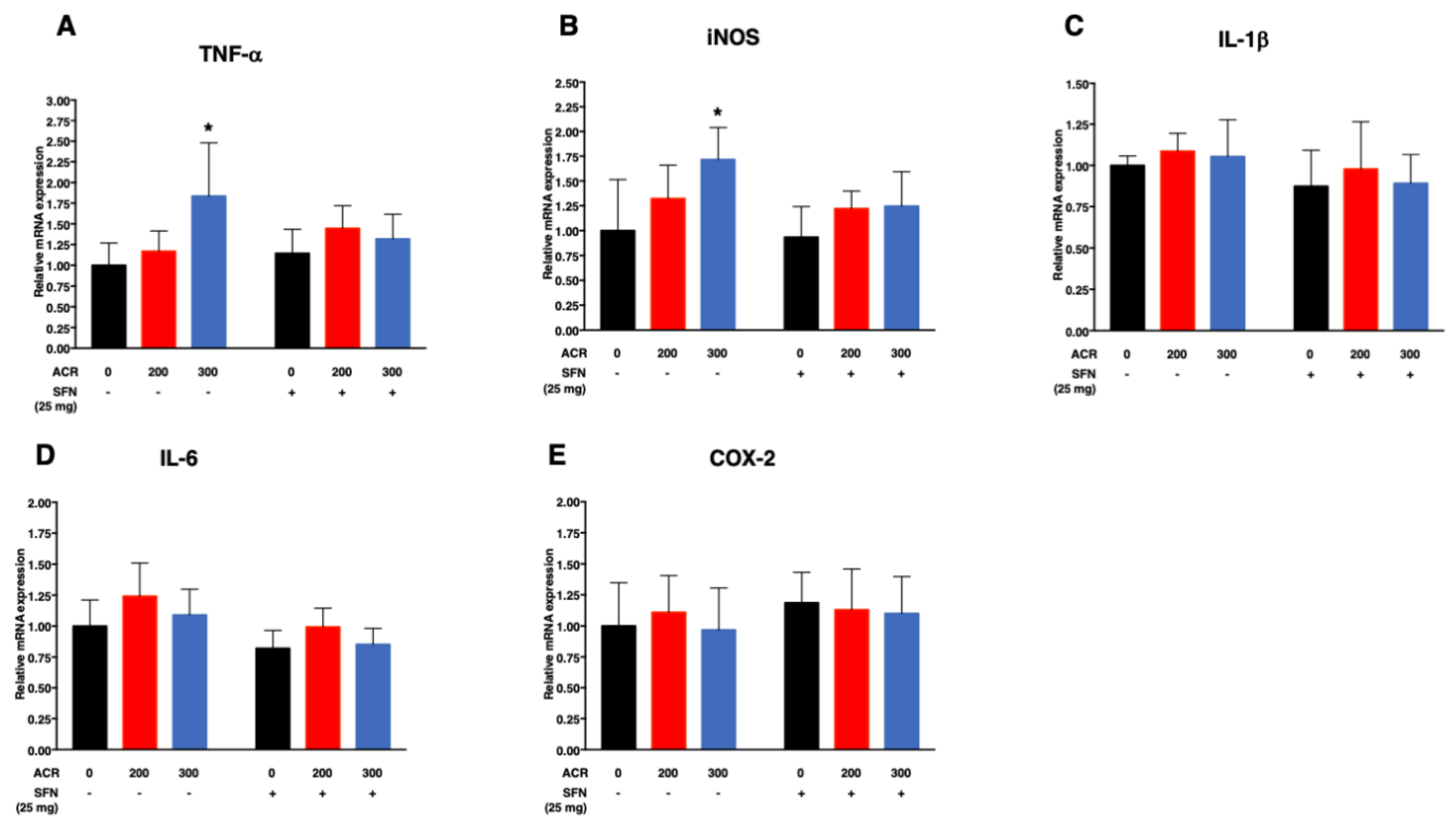
2.4.2. Pro-Inflammatory Cytokines
2.5. Changes in Glutathione and Malondialdehyde (MDA) Levels
2.6. Effects of Acrylamide on Liver Histopathology
3. Discussion
4. Methods
4.1. Chemicals and Preparation
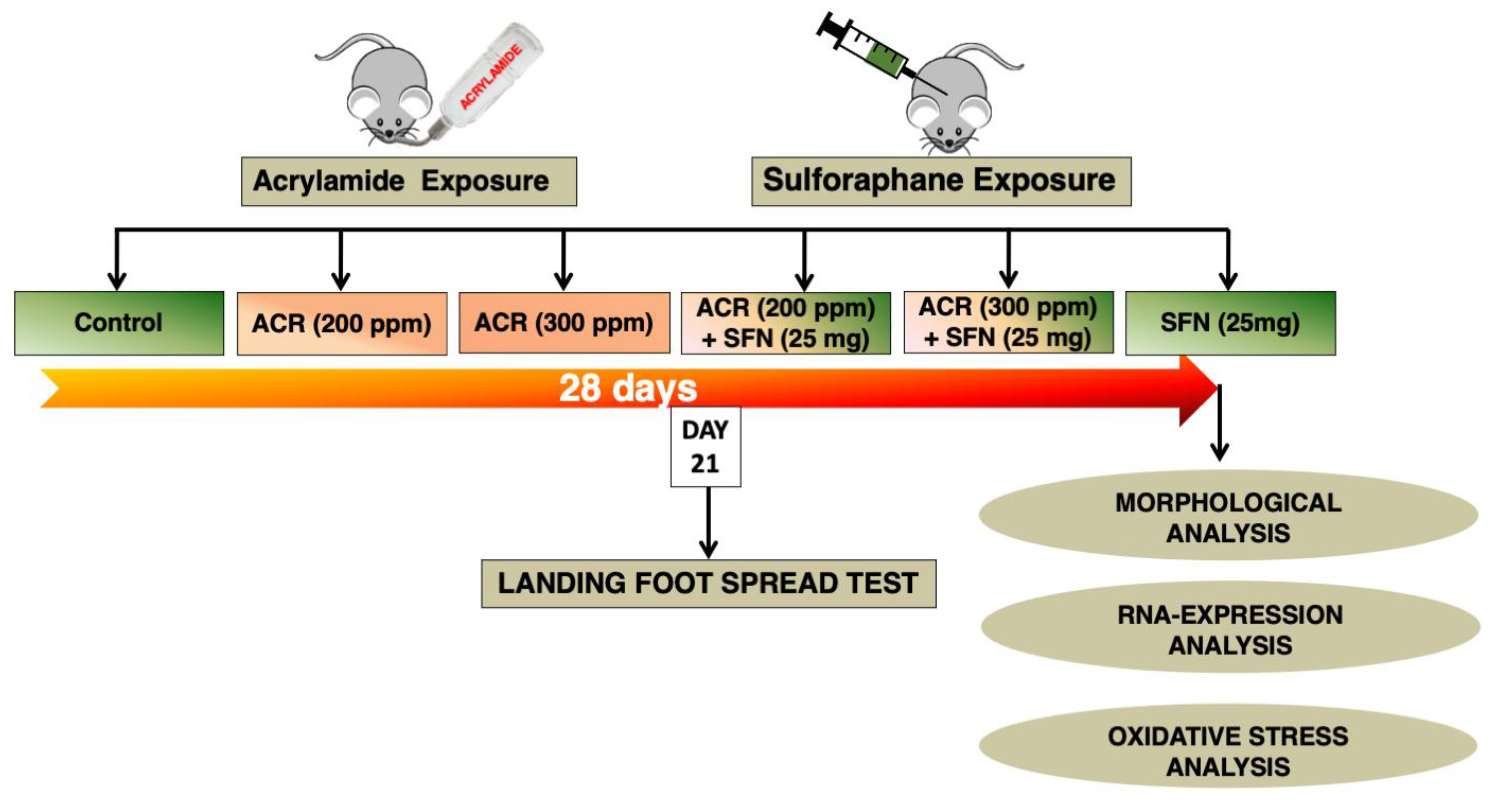
4.2. Animal Husbandry and Experimental Design
4.3. Concentration of Acrylamide
4.4. Amount of Acrylamide Uptake in Mice
4.5. Hindlimb Clasping Effect
4.6. Landing Foot Spread Test
4.7. Tissue Harvest, Processing and Morphological Assessment
Immunohistochemical Examination
4.8. Morphometric Analysis of Noradrenergic Axons
4.9. Tissue Processing and Histopathology
4.10. Tissue Harvest and Biochemical Assessment
4.11. Assessment of Oxidative Stress
4.11.1. Glutathione Assay (Quantification of Total and Oxidized Glutathione)
4.11.2. Thiobarbituric Acid Reactive Substances (TBARS) Assay
4.12. Total mRNA Isolation, cDNA Synthesis and Real-Time Quantitative Polymerase Chain Reaction (PCR)
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ruenz, M.; Bakuradze, T.; Eisenbrand, G.; Richling, E. Monitoring urinary mercapturic acids as biomarkers of human dietary exposure to acrylamide in combination with acrylamide uptake assessment based on duplicate diets. Arch. Toxicol. 2016, 90, 873–881. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T. Molecular mechanism of acrylamide neurotoxicity: Lessons learned from organic chemistry. Environ. Health Perspect. 2012, 120, 1650–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkekoğlu, P.; Baydar, T. Toxicity of acrylamide and evaluation of its exposure in baby foods. Nutr. Res. Rev. 2010, 23, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, P.S.; Schaumburg, H.H. A review of acrylamide neurotoxicity. Part II. Experimental animal neurotoxicity and pathologic mechanisms. Can. J. Neurol. Sci. 1974, 1, 152–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoPachin, R.M. The changing view of acrylamide neurotoxicity. Neurotoxicology 2004, 25, 617–630. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Ross, J.F.; Lehning, E.J. Nerve Terminals as the Primary Site of Acrylamide Action: A Hypothesis. Neurotoxicology 2002, 23, 43–59. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Ross, J.F.; Reid, M.L.; Das, S.; Mansukhani, S.; Lehning, E.J. Neurological Evaluation of Toxic Axonopathies in Rats: Acrylamide and 2,5-Hexanedione. Neurotoxicology 2002, 23, 95–110. [Google Scholar] [CrossRef]
- Dearfield, K.L.; Abernathy, C.O.; Ottley, M.S.; Brantner, J.H.; Hayes, P.F. Acrylamide: Its metabolism, developmental and reproductive effects, genotoxicity, and carcinogenicity. Mutat. Res. 1988, 195, 45–77. [Google Scholar] [CrossRef]
- Dearfield, K.L.; Douglas, G.R.; Ehling, U.H.; Moore, M.M.; Sega, G.A.; Brusick, D.J. Acrylamide: A review of its genotoxicity and an assessment of heritable genetic risk. Mutat. Res. 1995, 330, 71–99. [Google Scholar] [CrossRef]
- IARC. Monographs on the Evaluation of Carcinogenic Risks to Human: Some Industrial Chemicals; 60; International Agency for Research on Cancer: Lyon, France, 1994; pp. 389–433. [Google Scholar]
- Rice, J.M. The carcinogenicity of acrylamide. Mutat. Res. 2005, 580, 3–20. [Google Scholar] [CrossRef]
- Tyl, R.W.; Friedman, M.A. Effects of acrylamide on rodent reproductive performance. Reprod. Toxicol. 2003, 17, 1–13. [Google Scholar] [CrossRef]
- Smith, E.A.; Oehme, F.W. Acrylamide and polyacrylamide: A review of production, use, environmental fate and neurotoxicity. Rev. Environ. Health 1991, 9, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Bergmark, E. Hemoglobin adducts of acrylamide and acrylonitrile in laboratory workers, smokers and nonsmokers. Chem. Res. Toxicol. 1997, 10, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Mojska, H.; Gielecińska, I.; Cendrowski, A. Acrylamide content in cigarette mainstream smoke and estimation of exposure to acrylamide from tobacco smoke in Poland. Ann. Agric. Environ. Med. 2016, 23, 456–461. [Google Scholar] [CrossRef]
- Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S.; Törnqvist, M. Acrylamide: A cooking carcinogen? Chem. Res. Toxicol. 2000, 13, 517–522. [Google Scholar] [CrossRef]
- Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S.; Törnqvist, M. Analysis of acrylamide, a carcinogen formed in heated foodstuffs. J. Agric. Food Chem. 2002, 50, 4998–5006. [Google Scholar] [CrossRef]
- Lukac, H.; Amrein, T.M.; Perren, R.; Conde-Petit, B.; Amadò, R.; Escher, F. Influence of roasting conditions on the acrylamide content and the color of roasted almonds. J. Food Sci. 2007, 72, C033–C038. [Google Scholar] [CrossRef]
- Ishihara, K.; Matsunaga, A.; Miyoshi, T.; Nakamura, K.; Nakayama, T.; Ito, S.; Koga, H. Formation of acrylamide in a processed food model system, and examination of inhibitory conditions. Shokuhin Eiseigaku Zasshi 2005, 46, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Elmore, J.S.; Koutsidis, G.; Dodson, A.T.; Mottram, D.S.; Wedzicha, B.L. The effect of cooking on acrylamide and its precursors in potato, wheat and rye. Adv. Exp. Med. Biol. 2005, 561, 255–269. [Google Scholar] [CrossRef]
- Exon, J.H. A review of the toxicology of acrylamide. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 397–412. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small maf heterodimer mediates the induction of phase ii detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in keap1 are required for keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.-L.; Landar, A.; Ramachandran, A.; Ceaser, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Suzuki, H.; Itoh, K.; Yamamoto, M.; Sugiyama, Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein1 in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun. 2003, 310, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef] [Green Version]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y.H.; Hwang, Y.; Liu, H.; Wang, X.J.; Zhang, Y.; Stephenson, K.K.; Boronina, T.N.; Cole, R.N.; Dinkova-Kostova, A.T.; Talalay, P.; et al. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proc. Natl. Acad. Sci. USA 2010, 107, 9590–9595. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Ichihara, S.; Valentine, W.M.; Itoh, K.; Yamamoto, M.; Sheik Mohideen, S.; Kitoh, J.; Ichihara, G. Increased susceptibility of Nrf2-null mice to 1-bromopropane–induced hepatotoxicity. Toxicol. Sci. 2010, 115, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, T.; Shinkai, Y.; Jiang, H.-Y.; Iwamoto, N.; Sumi, D.; Taguchi, K.; Yamamoto, M.; Jinno, H.; Tanaka-Kagawa, T.; Cho, A.K.; et al. Initial response and cellular protection through the Keap1/Nrf2 system during the exposure of primary mouse hepatocytes to 1,2-naphthoquinone. Chem. Res. Toxicol. 2011, 24, 559–567. [Google Scholar] [CrossRef]
- Shinkai, Y.; Kimura, T.; Itagaki, A.; Yamamoto, C.; Taguchi, K.; Yamamoto, M.; Kumagai, Y.; Kaji, T. Partial contribution of the Keap1-Nrf2 system to cadmium-mediated metallothionein expression in vascular endothelial cells. Toxicol. Appl. Pharmacol. 2016, 295, 37–46. [Google Scholar] [CrossRef]
- Toyama, T.; Shinkai, Y.; Yasutake, A.; Uchida, K.; Yamamoto, M.; Kumagai, Y. Isothiocyanates reduce mercury accumulation via an nrf2-dependent mechanism during exposure of mice to methylmercury. Environ. Health Perspect. 2011, 119, 1117–1122. [Google Scholar] [CrossRef]
- Toyama, T.; Sumi, D.; Shinkai, Y.; Yasutake, A.; Taguchi, K.; Tong, K.I.; Yamamoto, M.; Kumagai, Y. Cytoprotective role of nrf2/keap1 system in methylmercury toxicity. Biochem. Biophys. Res. Commun. 2007, 363, 645–650. [Google Scholar] [CrossRef]
- Ekuban, F.A.; Zong, C.; Takikawa, M.; Morikawa, K.; Sakurai, T.; Ichihara, S.; Itoh, K.; Yamamoto, M.; Ohsako, S.; Ichihara, G. Genetic ablation of Nrf2 exacerbates neurotoxic effects of acrylamide in mice. Toxicology 2021, 456, 152785. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Kataoka, H.; Inose, Y.; Akaike, A.; Koyama, Y.; Kume, T. Neuroprotective effect of an Nrf2-ARE activator identified from a chemical library on dopaminergic neurons. Eur. J. Pharmacol. 2018, 818, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, T.; Mao, L.; Zhang, F. Sulforaphane protects against brain diseases: Roles of cytoprotective enzymes. Austin J. Cerebrovasc. Dis. Stroke 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of sulforaphane in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8637. [Google Scholar] [CrossRef]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Hrelia, S.; Angeloni, C.; Cantelli-Forti, G.; Hrelia, P. Sulforaphane as an inducer of glutathione prevents oxidative stress-induced cell death in a dopaminergic-like neuroblastoma cell line. J. Neurochem. 2009, 111, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [Green Version]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 2019, 28, 746–758.e744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denzer, I.; Münch, G.; Pischetsrieder, M.; Friedland, K. S-allyl-L-cysteine and isoliquiritigenin improve mitochondrial function in cellular models of oxidative and nitrosative stress. Food Chem. 2016, 194, 843–848. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [Green Version]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef]
- Lalonde, R.; Strazielle, C. Brain regions and genes affecting limb-clasping responses. Brain Res. Rev. 2011, 67, 252–259. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, E.; Niimi, K.; Itakura, C. Motor coordination impairment in aged heterozygous rolling Nagoya, Cav2.1 mutant mice. Brain Res. 2009, 1279, 50–57. [Google Scholar] [CrossRef]
- Kuperman, A.S. Effects of acrylamide on the central nervous system of the cat. J. Pharmacol. Exp. Ther. 1958, 123, 180–192. [Google Scholar]
- Leswing, R.J.; Ribelin, W.E. Physiologic and pathologic changes in acrylamide neuropathy. Arch. Environ. Health 1969, 18, 23–29. [Google Scholar] [CrossRef]
- Lowndes, H.E.; Baker, T.; Cho, E.S.; Jortner, B.S. Position sensitivity of de-efferented muscle spindles in experimental acrylamide neuropathy. J. Pharmacol. Exp. Ther. 1978, 205, 40–48. [Google Scholar] [PubMed]
- McCollister, D.D.; Oyen, F.; Rowe, V.K. Toxicology of Acrylamide. Toxicol. Appl. Pharmacol. 1964, 6, 172–181. [Google Scholar] [CrossRef]
- Fullerton, P.M.; Barnes, J.M. Peripheral neuropathy in rats produced by acrylamide. Br. J. Ind. Med. 1966, 23, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Pfaff, L.D. Acrylamide neuropathy in rats. An electron microscopic study of degeneration and regeneration. Acta Neuropathol. 1973, 24, 197–213. [Google Scholar] [CrossRef]
- Edwards, P.M. Neurotoxicity of acrylamide and its analogues and effects of these analogues and other agents on acrylamide neuropathy. Br. J. Ind. Med. 1975, 32, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Bradley, W.G.; Asbury, A.K. Radioautographic studies of Schwann cell behavior. I. Acrylamide neuropathy in the mouse. J. Neuropathol. Exp. Neurol. 1970, 29, 500–506. [Google Scholar] [CrossRef]
- Gilbert, S.G.; Maurissen, J.P.J. Assessment of the effects of acrylamide, methylmercury, and 2,5-hexanedione on motor functions in mice. J. Toxicol. Environ. Health 1982, 10, 31–41. [Google Scholar] [CrossRef]
- Hopkins, A. The effect of acrylamide on the peripheral nervous system of the baboon. J. Neurol. Neurosurg. Psychiatry 1970, 33, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.P.; Gilliatt, R.W. Motor and sensory nerve conduction velocity in the baboon: Normal values and changes during acrylamide neuropathy. J. Neurol. Neurosurg. Psychiatry 1971, 34, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Cabe, P.A.; Colwell, P.B. Toxic effects of acrylamide in Japanese quail (Coturnix coturnix Japonica). J. Toxicol. Environ. Health 1981, 7, 935–940. [Google Scholar] [CrossRef]
- Auld, R.B.; Bedwell, S.F. Peripheral neuropathy with sympathetic overactivity from industrial contact with acrylamide. Can. Med. Assoc. J. 1967, 96, 652–654. [Google Scholar] [PubMed]
- Garland, T.O.; Patterson, M.W. Six cases of acrylamide poisoning. Br. Med. J. 1967, 4, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Rho, H.J.; Kim, J.H.; Lee, S.H. Function of selective neuromodulatory projections in the mammalian cerebral cortex: Comparison between cholinergic and noradrenergic systems. Front. Neural Circuits 2018, 12, 47. [Google Scholar] [CrossRef]
- Kropf, E.; Syan, S.K.; Minuzzi, L.; Frey, B.N. From anatomy to function: The role of the somatosensory cortex in emotional regulation. Braz J. Psychiatry 2019, 41, 261–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bufalari, I.; Aprile, T.; Avenanti, A.; Di Russo, F.; Aglioti, S.M. Empathy for pain and touch in the human somatosensory cortex. Cereb. Cortex 2007, 17, 2553–2561. [Google Scholar] [CrossRef]
- Eickhoff, S.B.; Jbabdi, S.; Caspers, S.; Laird, A.R.; Fox, P.T.; Zilles, K.; Behrens, T.E. Anatomical and functional connectivity of cytoarchitectonic areas within the human parietal operculum. J. Neurosci. 2010, 30, 6409–6421. [Google Scholar] [CrossRef]
- Timmermann, L.; Ploner, M.; Haucke, K.; Schmitz, F.; Baltissen, R.; Schnitzler, A. Differential coding of pain intensity in the human primary and secondary somatosensory cortex. J. Neurophysiol. 2001, 86, 1499–1503. [Google Scholar] [CrossRef] [Green Version]
- Kaas, J.H.; Nelson, R.J.; Sur, M.; Lin, C.S.; Merzenich, M.M. Multiple representations of the body within the primary somatosensory cortex of primates. Science 1979, 204, 521–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbert, T.; Candia, V.; Altenmüller, E.; Rau, H.; Sterr, A.; Rockstroh, B.; Pantev, C.; Taub, E. Alteration of digital representations in somatosensory cortex in focal hand dystonia. Neuroreport 1998, 9, 3571–3575. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.; Premji, A.; Nelson, A.J. Plasticity-inducing TMS protocols to investigate somatosensory control of hand function. Neural Plast. 2012, 2012, 350574. [Google Scholar] [CrossRef]
- Konczak, J.; Abbruzzese, G. Focal dystonia in musicians: Linking motor symptoms to somatosensory dysfunction. Front. Hum. Neurosci. 2013, 7, 297. [Google Scholar] [CrossRef] [Green Version]
- Wolpert, D.; Pearson, K.; Ghez, C. The organization and planning of movement. In Principles of Neural Science, 5th ed.; Kandel, E., Schwartz, J., Jessell, T., Seigelbaum, S., Hudspeth, A., Eds.; McGraw-Hill Companies: New York, NY, USA, 2013; pp. 743–767. [Google Scholar]
- Hummelsheim, H.; Bianchetti, M.; Wiesendanger, M.; Wiesendanger, R. Sensory inputs to the agranular motor fields: A comparison between precentral, supplementary-motor and premotor areas in the monkey. Exp. Brain Res. 1988, 69, 289–298. [Google Scholar] [CrossRef]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Leoncini, E.; Malaguti, M.; Angelini, S.; Hrelia, P.; Hrelia, S. Modulation of phase II enzymes by sulforaphane: Implications for its cardioprotective potential. J. Agric. Food Chem. 2009, 57, 5615–5622. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S128–S138. [Google Scholar] [CrossRef]
- Vallee, B.L. Implications and inferences of metallothionein structure. Exp. Suppl. 1987, 52, 5–16. [Google Scholar] [CrossRef]
- Maret, W. Cellular zinc and redox states converge in the metallothionein/thionein pair. J. Nutr. 2003, 133, 1460s–1462s. [Google Scholar] [CrossRef] [Green Version]
- Maret, W. Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthy, N.; Shyam Sunder, A.; Manisenthil Kumar, K.; Senthil Kumar, M.; Ganesh, G.; Chatterjee, M. A review of metallothionein isoforms and their role in pathophysiology. World J. Surg. Oncol. 2011, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endresen, L.; Bakka, A.; Rugstad, H.E. Increased resistance to chlorambucil in cultured cells with a high concentration of cytoplasmic metallothionein. Cancer Res. 1983, 43, 2918–2926. [Google Scholar]
- Oh, S.H.; Deagen, J.T.; Whanger, P.D.; Weswig, P.H. Biological function of metallothionein. V. Its induction in rats by various stresses. Am. J. Physiol. 1978, 234, E282–E285. [Google Scholar] [CrossRef]
- Petering, D.H.; Krezoski, S.; Tabatabai, N.M. 12 Metallothionein toxicology: Metal ion trafficking and cellular protection metallothioneins and related chelators. In Metallothioneins and Related Chelators; Sigel, H., Sigel, A., Sigel, R.K.O., Eds.; De Gruyter: Berlin, Germany, 2009; Volume 48, pp. 353–398. [Google Scholar]
- Hao, Q.; Maret, W. Aldehydes release zinc from proteins. A pathway from oxidative stress/lipid peroxidation to cellular functions of zinc. FEBS J. 2006, 273, 4300–4310. [Google Scholar] [CrossRef]
- Ghorbel, I.; Elwej, A.; Chaabene, M.; Boudawara, O.; Marrakchi, R.; Jamoussi, K.; Boudawara, T.S.; Zeghal, N. Effects of acrylamide graded doses on metallothioneins I and II induction and DNA fragmentation: Bochemical and histomorphological changes in the liver of adult rats. Toxicol. Ind. Health 2017, 33, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Cheng, Y.; Wu, H.; Kong, L.; Wang, S.; Xu, Z.; Zhang, Z.; Tan, Y.; Keller, B.B.; Zhou, H.; et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes 2017, 66, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, Y.; Lou, Y.; Cui, W.; Miao, L. Sulforaphane suppresses obesity-related glomerulopathy-induced damage by enhancing autophagy via Nrf2. Life Sci. 2020, 258, 118153. [Google Scholar] [CrossRef]
- Wu, H.; Kong, L.; Cheng, Y.; Zhang, Z.; Wang, Y.; Luo, M.; Tan, Y.; Chen, X.; Miao, L.; Cai, L. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic. Biol. Med. 2015, 89, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; So, A.; Murphy, T.H. Activation of endogenous antioxidant defenses in neuronal cells prevents free radical-mediated damage. J. Neurochem. 1998, 71, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lee, J.M.; Johnson, J.A. Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J. Biol. Chem. 2002, 277, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.H.; De Long, M.J.; Coyle, J.T. Enhanced NAD(P)H:quinone reductase activity prevents glutamate toxicity produced by oxidative stress. J. Neurochem. 1991, 56, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Mas, S.; Gassó, P.; Trias, G.; Bernardo, M.; Lafuente, A. Sulforaphane protects SK-N-SH cells against antipsychotic-induced oxidative stress. Fundam. Clin. Pharmacol. 2012, 26, 712–721. [Google Scholar] [CrossRef]
- Mizuno, K.; Kume, T.; Muto, C.; Takada-Takatori, Y.; Izumi, Y.; Sugimoto, H.; Akaike, A. Glutathione biosynthesis via activation of the nuclear factor E2-related factor 2 (Nrf2)--antioxidant-response element (ARE) pathway is essential for neuroprotective effects of sulforaphane and 6-(methylsulfinyl) hexyl isothiocyanate. J. Pharmacol. Sci. 2011, 115, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.Y.; Ahmad, I.M.; Switzer, B.; Britigan, B.E. Induction of heme oxygenase-1 contributes to survival of Mycobacterium abscessus in human macrophages-like THP-1 cells. Redox Biol. 2015, 4, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prawan, A.; Kundu, J.K.; Surh, Y.J. Molecular basis of heme oxygenase-1 induction: Implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 2005, 7, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 induction in cancer progression: A matter of cell adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhäuser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, H.; Soroka, Y.; Zlotkin-Frušić, M.; Gilhar, A.; Milner, Y.; Tamir, S. Isothiocyanates inhibit psoriasis-related proinflammatory factors in human skin. Inflamm. Res. 2012, 61, 735–742. [Google Scholar] [CrossRef]
- Brandenburg, L.O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2010, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Folkard, D.L.; Melchini, A.; Traka, M.H.; Al-Bakheit, A.; Saha, S.; Mulholland, F.; Watson, A.; Mithen, R.F. Suppression of LPS-induced transcription and cytokine secretion by the dietary isothiocyanate sulforaphane. Mol. Nutr. Food Res. 2014, 58, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, L.; Chen, B.; Zheng, P.; He, Y.; Ding, Y.; Deng, Y.; Lu, X.; Guo, X.; Zhang, Y.; et al. DNA demethylation upregulated nrf2 expression in Alzheimer’s disease cellular model. Front. Aging Neurosci. 2015, 7, 244. [Google Scholar] [CrossRef] [Green Version]
- Parsanathan, R.; Jain, S.K. Glutathione deficiency induces epigenetic alterations of vitamin D metabolism genes in the livers of high-fat diet-fed obese mice. Sci. Rep. 2019, 9, 14784. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Manautou, J.E. Molecular mechanisms underlying chemical liver injury. Expert Rev. Mol. Med. 2012, 14, e4. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, B.; Kaplowitz, N. Clinical perspectives on xenobiotic-induced hepatotoxicity. Drug Metab. Rev. 2004, 36, 301–312. [Google Scholar] [CrossRef]
- Igisu, H.; Goto, I.; Kawamura, Y.; Kato, M.; Izumi, K. Acrylamide encephaloneuropathy due to well water pollution. J. Neurol. Neurosurg. Psychiatry 1975, 38, 581–584. [Google Scholar] [CrossRef]
- Morimoto, M. Occurence of human cases intoxicated with well water contaminated with acrylamide in Fukuoka prfecture. Water Waste 1975, 17, 51–62. [Google Scholar]
- Kesson, C.M.; Baird, A.W.; Lawson, D.H. Acrylamide poisoning. Postgrad. Med. J. 1977, 53, 16–17. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, D.; Bottani, E.; Segala, A.; Marchet, S.; Rossi, F.; Orlando, F.; Malavolta, M.; Carruba, M.O.; Lamperti, C.; Provinciali, M.; et al. Targeting multiple mitochondrial processes by a Metabolic Modulator prevents sarcopenia and cognitive decline in SAMP8 mice. Front. Pharmacol. 2020, 11, 1171. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Li, Y.F.; Wang, Z.T.; Jia, W.Q.; Xu, R.X. Toll-Like receptor 4 deficiency impairs motor coordination. Front. Neurosci. 2016, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Franklin, K.B.J. (Eds.) The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Elsevier Academic Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.M.; Wang, Y.; Tan, H.S.; Yu, T.; Fan, X.M.; Chen, P.; Zeng, H.; Huang, M.; Bi, H.C. Schisandrol B protects against acetaminophen-induced acute hepatotoxicity in mice via activation of the NRF2/ARE signaling pathway. Acta Pharmacol. Sin. 2016, 37, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Rojo de la Vega, M.; Quijada, H.; Wondrak, G.T.; Wang, T.; Garcia, J.G.N.; Zhang, D.D. Bixin protects mice against ventilation-induced lung injury in an NRF2-dependent manner. Sci. Rep. 2016, 6, 18760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, G.; Narasimhan, M.; Tamowski, S.; Darley-Usmar, V.; Rajasekaran, N.S. Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 2017, 12, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Kim, T.; Kim, M.; Kim, M.; Song, H.; Kim, T.S.; Seo, E.; Lee, S.H.; Kim, H.; Kim, S.K.; et al. Hippo-Foxa2 signaling pathway plays a role in peripheral lung maturation and surfactant homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, 7732–7737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; García, C.; Fernández-Ruiz, J.; Grochot–Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehsah, R.; Wu, W.; Ichihara, S.; Hashimoto, N.; Hasegawa, Y.; Zong, C.; Itoh, K.; Yamamoto, M.; Elsayed, A.A.; El-Bestar, S.; et al. Role of Nrf2 in inflammatory response in lung of mice exposed to zinc oxide nanoparticles. Part. Fibre Toxicol. 2019, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checker, R.; Patwardhan, R.S.; Sharma, D.; Menon, J.; Thoh, M.; Bhilwade, H.N.; Konishi, T.; Sandur, S.K. Schisandrin B exhibits anti-inflammatory activity through modulation of the redox-sensitive transcription factors Nrf2 and NF-κB. Free Radic. Biol. Med. 2012, 53, 1421–1430. [Google Scholar] [CrossRef]
- Kim, J.H.; Nam, Y.P.; Jeon, S.M.; Han, H.S.; Suk, K. Amyloid neurotoxicity is attenuated by metallothionein: Dual mechanisms at work. J. Neurochem. 2012, 121, 751–762. [Google Scholar] [CrossRef]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2009, 58, 588–598. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Parameters | Treatment | Concentration of Acrylamide (ppm) | Simple Regression | Multiple Regression (p Value) | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 200 | 300 | Regression Coefficient of ACR (p Value) | Interaction of ACR and SFN | Regression Coefficient of ACR | Regression Coefficient of SFN | ||
| Body Weight (g) | SFN (−) | 25.4 ± 1.2 | 24.8 ± 1.1 | 23.3 ± 1.5 * | −0.007 (0.002) g/ppm | 0.004 (0.17) | −0.007 (0.0005) g/ppm | 0.25 (0.42)/mg |
| SFN (+) | 25.1 ± 1.3 | 24.9 ± 0.8 | 24.1 ± 1.1 | −0.003 (0.07) g/ppm | ||||
| Landing foot spread (cm) | SFN (−) | 2.7 ± 0.4 | 3.7 ± 0.5 * | 4.5 ± 0.7 * | 0.006 (<0.0001) cm/ppm | −0.002 (0.12) | 0.006 (<0.0001) cm/ppm | −0.37 (0.02)/mg |
| SFN (+) | 2.5±0.5 | 3.6±0.3 * | 3.7±1.0 * | 0.004 (0.0003) cm/ppm | ||||
| Test Parameter | Region | Treatment | Simple Regression | Multiple Regression (p Value) | ||
|---|---|---|---|---|---|---|
| Regression Coefficient of ACR (p Value) | Interaction of ACR and SFN | Regression Coefficient of ACR | Regression Coefficient of SFN | |||
| Density of noradrenergic axons (%) | S1HL | SFN (−) | −0.03 (0.02) %/ppm | 0.02 (0.07) | −0.03 (0.001) %/ppm | 1.4 (0.3)/mg |
| SFN (+) | −0.008 (0.03) %/ppm | |||||
| S1BF | SFN (−) | −0.04 (0.001) %/ppm | −0.03 (0.009) | - | - | |
| SFN (+) | −0.007 (0.24) %/ppm | |||||
| S1FL | SFN (−) | −0.02 (0.01) %/ppm | 0.01 (0.23) | −0.02 (0.001) %/ppm | 0.41 (0.70)/mg | |
| SFN (+) | −0.01 (0.02) %/ppm | |||||
| S2 | SFN (−) | −0.04 (<0.0001) %/ppm | 0.03 (0.002) | - | - | |
| SFN (+) | −0.007 (0.29) %/ppm | |||||
| Test Parameters | Treatment | Simple Regression | Multiple Regression (p value) | ||
|---|---|---|---|---|---|
| Regression Coefficient of ACR (p Value) | Interaction of ACR and SFN | Regression Coefficient of ACR | Regression Coefficient of SFN | ||
| SOD-1 | SFN (−) | 0.0005 (0.02)/ppm | 0.0007 (0.09) | 0.0005 (0.10)/ppm | 0.26 (<0.0001)/mg |
| SFN (+) | 0.001 (0.005)/ppm | ||||
| NQO1 | SFN (−) | 0.002 (0.04)/ppm | 0.00008 (0.95) | 0.002 (0.02)/ppm | 0.33 (0.05)/mg |
| SFN (+) | 0.002 (0.02)/ppm | ||||
| HO-1 | SFN (−) | 0.002 (0.006)/ppm | −0.002 (0.06) | 0.002 (0.002)/ppm | -0.26 (0.02)/mg |
| SFN (+) | 0.0004 (0.45)/ppm | ||||
| GST-M5 | SFN (−) | 0.0006 (0.08)/ppm | 0.0003 (0.45) | 0.0006 (0.07)/ppm | 0.23 (0.0001)/mg |
| SFN (+) | 0.0009 (0.01)/ppm | ||||
| GST-M | SFN (−) | 0.001 (0.004)/ppm | 0.0003 (0.67) | 0.001 (0.003)/ppm | 0.26 (0.002)/mg |
| SFN (+) | 0.002 (0.002)/ppm | ||||
| NRF2 | SFN (−) | 0.0005 (0.02)/ppm | 0.0003 (0.33) | 0.0005 (0.05)/ppm | 0.07 (0.11)/mg |
| SFN (+) | 0.0008 (0.01)/ppm | ||||
| TXNRD1 | SFN (−) | −0.0001 (0.81)/ppm | 0.002 (0.004) | - | - |
| SFN (+) | 0.002 (<0.0001)/ppm | ||||
| MT-1 | SFN (−) | 0.003 (0.0008)/ppm | 0.002 (0.03) | - | - |
| SFN (+) | 0.005 (<0.0001)/ppm | ||||
| Test Parameters | Treatment | Simple Regression | Multiple Regression (p Value) | ||
|---|---|---|---|---|---|
| Regression Coefficient of ACR (p Value) | Interaction of ACR and SFN | Regression Coefficient of ACR | Regression Coefficient of SFN | ||
| TNF-α | SFN (−) | 0.003 (0.01)/ppm | −0.002 (0.09) | 0.003 (0.002)/ppm | −0.03 (0.80)/mg |
| SFN (+) | 0.0007 (0.22)/ppm | ||||
| iNOS | SFN (−) | 0.002 (0.008)/ppm | −0.001 (0.21) | 0.002 (0.001)/ppm | −0.21 (0.07)/mg |
| SFN (+) | 0.001 (0.05)/ppm | ||||
| IL-Iβ | SFN (−) | 0.0002 (0.45)/ppm | −0.00009 (0.86) | 0.0002 (0.56)/ppm | −0.13 (0.05)/mg |
| SFN (+) | 0.0001 (0.78)/ppm | ||||
| IL-6 | SFN (−) | 0.0004 (0.36)/ppm | −0.0002 (0.70) | 0.0004 (0.27)/ppm | −0.22 (0.003)/mg |
| SFN (+) | 0.0002 (0.47)/ppm | ||||
| COX-2 | SFN (−) | −0.00001 (0.99)/ppm | −0.0003 (0.74) | −0.00001 (0.98)/ppm | 0.11 (0.27)/mg |
| SFN (+) | −0.0003 (0.60)/ppm | ||||
| Test Parameters | Treatment | Acrylamide Concentration (ppm) | Simple Regression | Multiple Regression (p Value) | ||||
|---|---|---|---|---|---|---|---|---|
| 0 | 200 | 300 | ACR Regression Coefficient (p Value) | Interaction of ACR and SFN | ACR Regression Coefficient | SFN Regression Coefficient | ||
| Total Glutathione (GSH + GSSG, µM) | SFN (−) | 92.1 ± 12.3 | 94.4 ± 18.0 | 103.3 ± 21.3 | 0.034 (0.32)/ppm | −0.05 (0.29) | 0.03 (0.28)/ppm | 16.69 (0.004)/mg |
| SFN (+) | 115.7 ± 16.1 | 111.8 ± 6.7 | 112.2 ± 20.3 | −0.013 (0.66)/ppm | ||||
| Glutathione Disulfide (GSSG, µM) | SFN (−) | 2.7 ± 2.8 | 2.8 ± 2.2 | 3.7 ± 1.5 | 0.003 (0.46)/ppm | −0.004 (0.51) | 0.003 (0.45)/ppm | −0.36 (0.62)/mg |
| SFN (+) | 2.5 ± 1.4 | 3.6 ± 2.3 | 2.0 ± 2.4 | −0.001 (0.86)/ppm | ||||
| GSSG/GSH ratio (×10−2) | SFN (−) | 3.3 ± 3.7 | 3.2 ± 2.9 | 4.0 ± 2.4 | 0.002 (0.73)/ppm | −0.002 (0.73) | 0.002 (0.67)/ppm | −1.11 (0.19)/mg |
| SFN (+) | 2.1 ± 1.1 | 3.2 ± 2.0 | 1.8 ± 1.9 | −0.0003 (0.92)/ppm | ||||
| MDA (µM) | SFN (−) | 6.5 ± 2.2 | 6.6 ± 2.2 | 8.7 ± 1.1 | 0.006 (0.10)/ppm | −0.015 (0.003) | - | - |
| SFN (+) | 8.5 ± 1.6 | 7.0 ± 1.3 | 5.9 ± 1.6 * | −0.008 (0.008)/ppm | ||||
| Gene | PRIMER SEQUENCES | REFERENCES |
|---|---|---|
| Nfe2l2 (Nrf2) | F: CGAGATATACGCAGGAGAGGTAAGAR: GCTCGACAATGTTCTCCAGCTT | [125] |
| Keap-1 | F: GATCGGCTGCACTGAACTGR: GGCAGTGTGACAGGTTGAAG | [126] |
| Gst-M (GSTµ) | F: CTGAAGGTGGAATACTTGGAGCR: GCCCAGGAACTGTGAGAAGA | [127] |
| GST-M5 | F: AGAAACGGTACATCTGTGGGGR: GGATGGCGTTACTCTGGGTG | [128] |
| HO-1 | F: CACAGATGGCGTCACTTCCGTCR: GTGAGGACCCACTGGGAGGAG | [129] |
| NQO1 | F: AGCGTTCGGTATTACGATCCR: AGTACAATCAGGGCTCTTCTCG | [130] |
| SOD-1 | F: CAGGACCTCATTTTAATCCTCACR: TGCCCAGGTCTCCAACAT | |
| TXNDR1 | F: GGGTCCTATGACTTCGACCTGR: AGTCGGTGTGACAAAATCCAAG | [131] |
| MT-1 | F: ACCTCCTGCAAGAAGAGCTGR: GCTGGGTTGGTCCGATACTA | [132] |
| TNF-α | F: CATCTTCTCAAAATTCGAGTGACAAR: TGGGAGTAGACAAGGTACAACCC | [36] |
| iNOS | F: CCTCCTTTGCCTCTCACTCTTR: AGTATTAGACGCGTGGCATGG | [133] |
| IL-1β | F: CTGGTGTGTGACGTTCCCATTAR: CCGACAGCACGAGGCTTT | |
| IL-6 | F: CCTACCCCAATTTCCAATGCTR: TATTTTCTGACCACAGTGAGGAAT | |
| COX-2 | F: TTCGGGAGCACAACAGAGTR: TAACCGCTCAGGTGTTGCAC | [36] |
| β-ACTIN | F: TCCTTCCTGGGCATGGAGR: AGGAGGAGCAATGATCTTGATCTT | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davuljigari, C.B.; Ekuban, F.A.; Zong, C.; Fergany, A.A.M.; Morikawa, K.; Ichihara, G. Nrf2 Activation Attenuates Acrylamide-Induced Neuropathy in Mice. Int. J. Mol. Sci. 2021, 22, 5995. https://doi.org/10.3390/ijms22115995
Davuljigari CB, Ekuban FA, Zong C, Fergany AAM, Morikawa K, Ichihara G. Nrf2 Activation Attenuates Acrylamide-Induced Neuropathy in Mice. International Journal of Molecular Sciences. 2021; 22(11):5995. https://doi.org/10.3390/ijms22115995
Chicago/Turabian StyleDavuljigari, Chand Basha, Frederick Adams Ekuban, Cai Zong, Alzahraa A. M. Fergany, Kota Morikawa, and Gaku Ichihara. 2021. "Nrf2 Activation Attenuates Acrylamide-Induced Neuropathy in Mice" International Journal of Molecular Sciences 22, no. 11: 5995. https://doi.org/10.3390/ijms22115995
APA StyleDavuljigari, C. B., Ekuban, F. A., Zong, C., Fergany, A. A. M., Morikawa, K., & Ichihara, G. (2021). Nrf2 Activation Attenuates Acrylamide-Induced Neuropathy in Mice. International Journal of Molecular Sciences, 22(11), 5995. https://doi.org/10.3390/ijms22115995