
Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies

 , and
, and 
Abstract
:1. Clinical Features, Classification and Genetics of CMT Neuropathies
2. Overview of Therapeutic Approaches for CMT Neuropathies
2.1. Gene Therapy
2.2. Small Molecule Therapies
3. Emerging Treatments for Demyelinating CMT Neuropathies
3.1. CMT1A
3.1.1. Gene Therapy Approaches for CMT1A
3.1.2. Drug-Based Therapies for CMT1A
3.2. CMT1B
Drug-Based Therapies for CMT1B
3.3. CMT1X
3.3.1. Gene Therapy for CMT1X
3.3.2. Targeting Inflammatory Pathways to Treat CMT1X
3.4. Other Demyelinating and Recessively Inherited CMT Forms
3.4.1. CMT4B
3.4.2. CMT4C
3.4.3. CMT4J
4. Treatment Approaches for Axonal CMT Types
4.1. CMT2A
4.1.1. Targeting the SARM1 Pathway
4.1.2. Agonists and Activators of MFN2 Mitochondrial Function
4.2. Distal Hereditary Motor Neuropathy (dHMN) Associated with SORD Gene Mutations
4.3. CMT2D
4.4. CMT2E
4.5. CMT2F
4.6. CMT2S
5. Clinical Trial Readiness for CMT Neuropathies
5.1. Clinical Evaluation Tools
5.2. MRI and Other Biomarkers
5.3. Optimal Trial Design for CMT Neuropathies
6. Summary and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kleopa, K.A.; Scherer, S.S. Inherited neuropathies. Neurol. Clin. 2002, 20, 679–709. [Google Scholar] [CrossRef]
- Baets, J.; De Jonghe, P.; Timmerman, V. Recent advances in Charcot-Marie-Tooth disease. Curr. Opin. Neurol. 2014, 27, 532–540. [Google Scholar] [CrossRef]
- Shy, M.E.; Lupski, J.R.; Chance, P.F.; Klein, C.J.; Dyck, P.J. Hereditary motor and sensory neuropathies: An overview of clinical, genetic, electrophysiologic, and pathologic features. In Peripheral Neuropathy; Elsevier BV, Elsivier Saunders: Philadelphia, PA, USA, 2005; pp. 1623–1658. [Google Scholar]
- Scherer, S.S.; Kleopa, K.A.; Benson, M.D. Peripheral Neuropathies. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease; Rosenberg, N., Pascual, J.M., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; pp. 345–365. [Google Scholar]
- Bird, T.D. Charcot-Marie-Tooth (CMT) hereditary neuropathy overview. In GeneReviews((R)); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Fridman, V.; Bundy, B.; Reilly, M.M.; Pareyson, D.; Bacon, C.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: A cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 873–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Laura, M.; Fawcett, K.; Pandraud, A.; Liu, Y.-T.; Davidson, G.L.; Rossor, A.M.; Polke, J.M.; Castleman, V.; Manji, H.; et al. Charcot–Marie–Tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 2012, 83, 706–710. [Google Scholar] [CrossRef]
- Saporta, A.S.; Ba, S.L.S.; Ms, L.J.M.; Ms, S.M.F.; Ms, C.E.S.; Shy, M.E. Charcot-marie-tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef]
- Wilmshurst, J.M.; Ouvrier, R. Hereditary peripheral neuropathies of childhood: An overview for clinicians. Neuromuscul. Disord. 2011, 21, 763–775. [Google Scholar] [CrossRef]
- Tao, F.; Beecham, G.W.; Rebelo, A.P.; Blanton, S.H.; Moran, J.J.; Lopez-Anido, C.; Svaren, J.; Abreu, L.; Rizzo, D.; Kirk, C.A.; et al. Modifier Gene Candidates in Charcot-Marie-Tooth Disease Type 1A: A Case-Only Genome-Wide Association Study. J. Neuromuscul. Dis. 2019, 6, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansagi, B.; Griffin, H.R.; Whittaker, R.; Antoniadi, T.; Evangelista, T.; Miller, J.; Greenslade, M.; Forester, N.; Duff, J.; Bradshaw, A.; et al. Genetic heterogeneity of motor neuropathies. Neurology 2017, 88, 1226–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotthier, A.; Baets, J.; De Vriendt, E.; Jacobs, A.; Auer-Grumbach, M.; Lévy, N.; Bonello-Palot, N.; Kilic, S.S.; Weis, J.; Nascimento, A.; et al. Genes for hereditary sensory and autonomic neuropathies: A genotype-phenotype correlation. Brain 2009, 132, 2699–2711. [Google Scholar] [CrossRef] [Green Version]
- Rotthier, A.; Baets, J.; Timmerman, V.; Janssens, K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 2012, 8, 73–85. [Google Scholar] [CrossRef]
- Mathis, S.; Tazir, M.; Solé, G.; Magy, L.; Le Masson, G.; Couratier, P.; Ghorab, K.; Duval, F.; Lacoste, I.; Goizet, C.; et al. Some new proposals for the classification of inherited myopathies. J. Neurol. Sci. 2018, 391, 118–119. [Google Scholar] [CrossRef]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef]
- Scherer, S.S.; Wrabetz, L. Molecular mechanisms of inherited demyelinating neuropathies. Glia 2008, 56, 1578–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargiannidou, I.; Kagiava, A.; Kleopa, K.A. Gene therapy approaches targeting Schwann cells for demyelinating neuropathies. Brain Res. 2020, 1728, 146572. [Google Scholar] [CrossRef]
- Stavrou, M.; Sargiannidou, I.; Christofi, T.; Kleopa, K.A. Genetic mechanisms of peripheral nerve disease. Neurosci. Lett. 2021, 742, 135357. [Google Scholar] [CrossRef]
- Kantor, B.; Bailey, R.M.; Wimberly, K.; Kalburgi, S.N.; Gray, S.J. Methods for Gene Transfer to the Central Nervous System. Adv. Genet. 2014, 87, 125–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Carroll, S.J.; Cook, W.H.; Young, D. AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Front. Mol. Neurosci. 2021, 13, 618020. [Google Scholar] [CrossRef]
- Sargiannidou, I.; Kagiava, A.; Bashiardes, S.; Richter, J.; Christodoulou, C.; Scherer, S.S.; Kleopa, K.A. IntraneuralGJB1gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 2015, 78, 303–316. [Google Scholar] [CrossRef]
- Kagiava, A.; Karaiskos, C.; Richter, J.; Tryfonos, C.; Lapathitis, G.; Sargiannidou, I.; Christodoulou, C.; Kleopa, K.A. Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum. Mol. Genet. 2018, 27, 1460–1473. [Google Scholar] [CrossRef] [PubMed]
- Kagiava, A.; Richter, J.; Tryfonos, C.; Karaiskos, C.; Heslegrave, A.J.; Sargiannidou, I.; Rossor, A.M.; Zetterberg, H.; Reilly, M.M.; Christodoulou, C.; et al. Gene replacement therapy after neuropathy onset provides therapeutic benefit in a model of CMT1X. Hum. Mol. Genet. 2019, 28, 3528–3542. [Google Scholar] [CrossRef] [PubMed]
- Kagiava, A.; Sargiannidou, I.; Theophilidis, G.; Karaiskos, C.; Richter, J.; Bashiardes, S.; Schiza, N.; Nearchou, M.; Christodoulou, C.; Scherer, S.S.; et al. Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. USA 2016, 113, E2421–E2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiza, N.; Georgiou, E.; Kagiava, A.; Médard, J.-J.; Richter, J.; Tryfonos, C.; Sargiannidou, I.; Heslegrave, A.J.; Rossor, A.M.; Zetterberg, H.; et al. Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy. Brain 2019, 142, 1227–1241. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kwak, G.; Kim, H.J.; Park, H.-T.; Choi, B.-O.; Bin Hong, Y. miR-381 Attenuates Peripheral Neuropathic Phenotype Caused by Overexpression of PMP22. Exp. Neurobiol. 2019, 28, 279–288. [Google Scholar] [CrossRef]
- Palfi, S.; Gurruchaga, J.M.; Ralph, G.S.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahenk, Z.; Galloway, G.; Clark, K.R.; Malik, V.; Rodino-Klapac, L.R.; Kaspar, B.K.; Chen, L.; Braganza, C.; Montgomery, C.; Mendell, J.R. AAV1.NT-3 gene therapy for charcot-marie-tooth neuropathy. Mol. Ther. 2014, 22, 511–521. [Google Scholar] [CrossRef]
- Serfecz, J.; Bazick, H.; Al Salihi, M.O.; Turner, P.; Fields, C.; Cruz, P.; Renne, R.; Notterpek, L. Downregulation of the human peripheral myelin protein 22 gene by miR-29a in cellular models of Charcot–Marie–Tooth disease. Gene Ther. 2019, 26, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, K.H.; Griffin, L.B.; Pyne, N.K.; Wallace, L.M.; Fowler, A.M.; Oprescu, S.N.; Takase, R.; Wei, N.; Meyer-Schuman, R.; Mellacheruvu, D.; et al. Allele-specific RNA interference prevents neuropathy in Charcot-Marie-Tooth disease type 2D mouse models. J. Clin. Investig. 2019, 129, 5568–5583. [Google Scholar] [CrossRef] [Green Version]
- Sahenk, Z.; Ozes, B. Gene therapy to promote regeneration in Charcot-Marie-Tooth disease. Brain Res. 2020, 1727, 146533. [Google Scholar] [CrossRef]
- Ozes, B.; Myers, M.; Moss, K.; Mckinney, J.; Ridgley, A.; Chen, L.; Bai, S.; Abrams, C.K.; Freidin, M.M.; Mendell, J.R.; et al. AAV1.NT-3 gene therapy for X-linked Charcot-Marie-Tooth neuropathy type 1. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Kagiava, A.; Karaiskos, C.; Richter, J.; Tryfonos, C.; Jennings, M.J.; Heslegrave, A.J.; Sargiannidou, I.; Stavrou, M.; Zetterberg, H.; Reilly, M.M.; et al. AAV9-mediated Schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther. 2021, 1–17. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Thenmozhi, R.; Lee, J.-S.; Park, N.Y.; Choi, B.-O.; Bin Hong, B.-O.C.A.Y. Gene Therapy Options as New Treatment for Inherited Peripheral Neuropathy. Exp. Neurobiol. 2020, 29, 177–188. [Google Scholar] [CrossRef]
- Pantera, H.; Moran, J.J.; Hung, H.A.; Pak, E.; Dutra, A.; Svaren, J. Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer. Hum. Mol. Genet. 2018, 27, 2830–2839. [Google Scholar] [CrossRef]
- Knauert, M.P.; Glazer, P.M. Triplex forming oligonucleotides: Sequence-specific tools for gene targeting Human. Mol. Genet. 2001, 10, 2243–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Classification and Nomenclature of CRISPR-Cas Systems: Where from Here? CRISPR J. 2018, 1, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Smargon, A.A.; Shi, Y.J.; Yeo, G.W. RNA-targeting CRISPR systems from metagenomic discovery to transcriptomic engineering. Nat. Cell Biol. 2020, 22, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Lee, J.Y.; Song, D.W.; Bae, H.S.; Doo, H.M.; Yu, H.S.; Lee, K.J.; Kim, H.K.; Hwang, H.; Kwak, G.; et al. Targeted PMP22 TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res. 2019, 48, 130–140. [Google Scholar] [CrossRef]
- Gautier, B.; Hajjar, H.; Soares, S.; Berthelot, J.; Deck, M.; Abbou, S.; Campbell, G.; Ceprian, M.; Gonzalez, S.; Fovet, C.-M.; et al. AAV2/9-mediated silencing of PMP22 prevents the development of pathological features in a rat model of Charcot-Marie-Tooth disease 1 A. Nat Commun. 2021, 12, 2356. [Google Scholar] [CrossRef]
- Sahenk, Z.; Nagaraja, H.N.; McCracken, B.S.; King, W.M.; Freimer, M.L.; Cedarbaum, J.M.; Mendell, J.R. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 2005, 65, 681–689. [Google Scholar] [CrossRef]
- Boutary, S.; Caillaud, M.; El Madani, M.; Vallat, J.-M.; Loisel-Duwattez, J.; Rouyer, A.; Richard, L.; Gracia, C.; Urbinati, G.; Desmaële, D.; et al. Squalenoyl siRNA PMP22 nanoparticles are effective in treating mouse models of Charcot-Marie-Tooth disease type 1 A. Commun. Biol. 2021, 4, 1–14. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Rizzo, F.; Salani, S.; Dametti, S.; Rinchetti, P.; Del Bo, R.; Foust, K.; Kaspar, B.K.; Bresolin, N.; et al. Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci. Adv. 2015, 1, e1500078. [Google Scholar] [CrossRef] [Green Version]
- Knabel, M.K.; Ramachandran, K.; Karhadkar, S.; Hwang, H.-W.; Creamer, T.J.; Chivukula, R.R.; Sheikh, F.; Clark, K.R.; Torbenson, M.; Montgomery, R.A.; et al. Systemic Delivery of scAAV8-Encoded MiR-29a Ameliorates Hepatic Fibrosis in Carbon Tetrachloride-Treated Mice. PLoS ONE 2015, 10, e0124411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Huang, S.X.; Strickland, A.; Doan, R.A.; Summers, D.W.; Mao, X.; Park, J.; DiAntonio, A.; Milbrandt, J. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 2019, 216, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Sancho, S.; Magyar, J.P.; Aguzzi, A.; Suter, U. Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain 1999, 122, 1563–1577. [Google Scholar] [CrossRef] [Green Version]
- Moss, K.R.; Höke, A. Targeting the programmed axon degeneration pathway as a potential therapeutic for Charcot-Marie-Tooth disease. Brain Res. 2020, 1727, 146539. [Google Scholar] [CrossRef]
- Rossaert, E.; Bosch, L.V.D. HDAC6 inhibitors: Translating genetic and molecular insights into a therapy for axonal CMT. Brain Res. 2020, 1733, 146692. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Bolhuis, P.A.; Zorn, I.; Hoogendijk, J.E.; van den Bosch, N.; Hensels, G.W.; Stanton, V.P., Jr.; Housman, D.E.; Fischbeck, K.H.; Ross, D.A.; et al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992, 1, 166–170. [Google Scholar] [CrossRef]
- Timmerman, V.; Nelis, E.; Van Hul, W.; Nieuwenhuijsen, B.; Chen, K.; Wang, S.; Ben Othman, K.; Cullen, B.; Leach, R.; Hanemann, C.O.; et al. The peripheral myelin protein gene PMP–22 is contained within the Charcot–Marie–Tooth disease type 1A duplication. Nat. Genet. 1992, 1, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, N.; Smith, B.; Ballard, L.; Lensch, M.W.; Robertson, M.; Albertsen, H.; Hanemann, C.O.; Müller, H.W.; Bird, T.D.; White, R.; et al. Peripheral myelin protein–22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot–Marie–Tooth 1A. Nat. Genet. 1992, 1, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Kiyosawa, H.; Lensch, M.; Chance, P.F. Analysis of the CMT1A-REP repeat: Mapping crossover breakpoints in CMT1A and HNPP. Hum. Mol. Genet. 1995, 4, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Snipes, G.J.; Suter, U.; Welcher, A.A.; Shooter, E.M. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J. Cell Biol. 1992, 117, 225–238. [Google Scholar] [CrossRef]
- Adlkofer, K.; Martini, R.; Aguzzi, A.; Zielasek, J.; Toyka, K.V.; Suter, U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat. Genet. 1995, 11, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.-G.; Ichihara, N.; Saigoh, K.; Nakabayashi, O.; Yamanishi, T.; Tanaka, K.; Wada, K.; Kikuchi, T. An in-frame deletion in peripheral myelin protein-22 gene causes hypomyelination and cell death of the schwann cells in the new Trembler mutant mice. Neuroscience 1997, 79, 735–744. [Google Scholar] [CrossRef]
- Fledrich, R.; Stassart, R.M.; Klink, A.; Rasch, L.M.; Prukop, T.; Haag, L.; Czesnik, D.; Kungl, T.; Abdelaal, T.A.M.; Keric, N.; et al. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat. Med. 2014, 20, 1055–1061. [Google Scholar] [CrossRef]
- Notterpek, L.; Roux, K.J.; Amici, S.A.; Yazdanpour, A.; Rahner, C.; Fletcher, B.S. Peripheral myelin protein 22 is a constituent of intercellular junctions in epithelia. Proc. Natl. Acad. Sci. USA 2001, 98, 14404–14409. [Google Scholar] [CrossRef] [Green Version]
- Sereda, M.W.; Nave, K.-A. Animal models of Charcot-Marie-Tooth disease type 1A. NeuroMolecular Med. 2006, 8, 205–215. [Google Scholar] [CrossRef]
- Perea, J.; Robertson, A.; Tolmachova, T.; Muddle, J.; King, R.H.M.; Ponsford, S.; Thomas, P.K.; Huxley, C. Induced myelination and demyelination in a conditional mouse model of Charcot-Marie-Tooth disease type 1A. Hum. Mol. Genet. 2001, 10, 1007–1018. [Google Scholar] [CrossRef] [Green Version]
- Chumakov, I.; Milet, A.; Cholet, N.; Primas, G.; Boucard, A.; Pereira, Y.; Graudens, E.; Mandel, J.; Laffaire, J.; Foucquier, J.; et al. Polytherapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet J. Rare Dis. 2014, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Prukop, T.; Stenzel, J.; Wernick, S.; Kungl, T.; Mroczek, M.; Adam, J.; Ewers, D.; Nabirotchkin, S.; Nave, K.-A.; Hajj, R.; et al. Early short-term PXT3003 combinational therapy delays disease onset in a transgenic rat model of Charcot-Marie-Tooth disease 1A (CMT1A). PLoS ONE 2019, 14, e0209752. [Google Scholar] [CrossRef] [PubMed]
- Prukop, T.; Wernick, S.; Boussicault, L.; Ewers, D.; Jäger, K.; Adam, J.; Winter, L.; Quintes, S.; Linhoff, L.; Barrantes-Freer, A.; et al. Synergistic PXT3003 therapy uncouples neuromuscular function from dysmyelination in male Charcot–Marie–Tooth disease type 1A (CMT1A) rats. J. Neurosci. Res. 2020, 98, 1933–1952. [Google Scholar] [CrossRef]
- Chahbouni, M.; Escames, G.; Venegas, C.; Sevilla, B.; García, J.A.; Lopez, L.C.; Muñoz-Hoyos, A.; Molina-Carballo, A.; Acuña-Castroviejo, D. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophy. J. Pineal Res. 2010, 48, 282–289. [Google Scholar] [CrossRef]
- Fledrich, R.; Abdelaal, T.; Rasch, L.; Bansal, V.; Schütza, V.; Brügger, B.; Lüchtenborg, C.; Prukop, T.; Stenzel, J.; Rahman, R.U.; et al. Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küllenberg, D.; Taylor, L.A.; Schneider, M.; Massing, U. Health effects of dietary phospholipids. Lipids Heal. Dis. 2012, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontés, M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat. Med. 2004, 10, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Ouvrier, R.A.; Yiu, E.M.; Joseph, P.D.; Kornberg, A.J.; Fahey, M.C.; Ryan, M.M. Ascorbic acid for Charcot–Marie–Tooth disease type 1A in children: A randomised, double-blind, placebo-controlled, safety and efficacy trial. Lancet Neurol. 2009, 8, 537–544. [Google Scholar] [CrossRef]
- Verhamme, C.; de Haan, R.J.; Vermeulen, M.; Baas, F.; de Visser, M.; van Schaik, I.N. Oral high dose ascorbic acid treatment for one year in young CMT1A patients: A randomised, double-blind, placebo-controlled phase II trial. BMC Med. 2009, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Pareyson, D.; Reilly, M.M.; Schenone, A.; Fabrizi, G.M.; Cavallaro, T.; Santoro, L.; Vita, G.; Quattrone, A.; Padua, L.; Gemignani, F.; et al. Ascorbic acid in Charcot–Marie–Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): A double-blind randomised trial. Lancet Neurol. 2011, 10, 320–328. [Google Scholar] [CrossRef]
- Lewis, R.A.; McDermott, M.P.; Herrmann, D.N.; Hoke, A.; Clawson, L.L.; Siskind, C.; Feely, S.M.E.; Miller, L.J.; Barohn, R.J.; Smith, P.; et al. Muscle Study Group, High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: Results of a randomized, double-masked, controlled trial. JAMA Neurol. 2013, 70, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Henry, T.D.; Hirsch, A.T.; Goldman, J.J.; Wang, Y.L.; Lips, D.L.; McMillan, W.D.; Duval, S.; Biggs, T.A.; Keo, H.H. Safety of a non-viral plasmid-encoding dual isoforms of hepatocyte growth factor in critical limb ischemia patients: A phase I study. Gene Ther. 2011, 18, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.R.; Lee, J.; Lee, D.; Nho, B.; Kim, S. Hepatocyte Growth Factor (HGF) Promotes Peripheral Nerve Regeneration by Activating Repair Schwann Cells. Sci. Rep. 2018, 8, 8316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Chang, E.H.; Koo, O.J.; Jwa, D.H.; Mo, W.M.; Kwak, G.; Moon, H.W.; Park, H.T.; Bin Hong, Y.; Choi, B.-O. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol. Dis. 2017, 100, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Nobbio, L.; Sturla, L.; Fiorese, F.; Usai, C.; Basile, G.; Moreschi, I.; Benvenuto, F.; Zocchi, E.; De Flora, A.; Schenone, A.; et al. P2X7-mediated Increased Intracellular Calcium Causes Functional Derangement in Schwann Cells from Rats with CMT1A Neuropathy. J. Biol. Chem. 2009, 284, 23146–23158. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Karli, I.-L.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J. Clin. Investig. 2017, 128, 359–368. [Google Scholar] [CrossRef]
- Hai, M.; Bidichandani, S.I.; Hogan, M.E.; Patel, P.I. Competitive Binding of Triplex-Forming Oligonucleotides in the Two Alternate Promoters of thePMP22Gene. Antisense Nucleic Acid Drug Dev. 2001, 11, 233–246. [Google Scholar] [CrossRef]
- Ha, N.; Choi, Y.I.; Jung, N.; Song, J.Y.; Bae, D.K.; Kim, M.C.; Lee, Y.J.; Song, H.; Kwak, G.; Jeong, S.; et al. A novel histone deacetylase 6 inhibitor improves myelination of Schwann cells in a model of Charcot–Marie–Tooth disease type 1A. Br. J. Pharmacol. 2020, 177, 5096–5113. [Google Scholar] [CrossRef]
- Khajavi, M.; Shiga, K.; Wiszniewski, W.; He, F.; Shaw, C.A.; Yan, J.; Wensel, T.G.; Snipes, G.J.; Lupski, J.R. Oral Curcumin Mitigates the Clinical and Neuropathologic Phenotype of the Trembler-J Mouse: A Potential Therapy for Inherited Neuropathy. Am. J. Hum. Genet. 2007, 81, 438–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntoutoume, G.M.N.; Granet, R.; Mbakidi, J.P.; Brégier, F.; Léger, D.Y.; Fidanzi-Dugas, C.; Lequart, V.; Joly, N.; Liagre, B.; Chaleix, V.; et al. Development of curcumin–cyclodextrin/cellulose nanocrystals complexes: New anticancer drug delivery systems. Bioorganic Med. Chem. Lett. 2016, 26, 941–945. [Google Scholar] [CrossRef]
- Sereda, M.W.; Zu Hörste, G.M.; Suter, U.; Uzma, N.; Nave, K.-A. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat. Med. 2003, 9, 1533–1537. [Google Scholar] [CrossRef]
- Zu Horste, G.M.; Prukop, T.; Liebetanz, D.; Möbius, W.; Nave, K.-A.; Sereda, M.W. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann. Neurol. 2007, 61, 61–72. [Google Scholar] [CrossRef]
- Fledrich, R.; Akkermann, D.; Schütza, V.; Abdelaal, T.A.; Hermes, D.; Schäffner, E.; Soto-Bernardini, M.C.; Götze, T.; Klink, A.; Kusch, K.; et al. NRG1 type I dependent autoparacrine stimulation of Schwann cells in onion bulbs of peripheral neuropathies. Nat. Commun. 2019, 10, 1840. [Google Scholar] [CrossRef] [PubMed]
- Madorsky, I.; Opalach, K.; Waber, A.; Verrier, J.D.; Solmo, C.; Foster, T.; Dunn, W.A., Jr.; Notterpek, L. Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol. of Dis. 2009, 34, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, S.; Verrier, J.D.; Madorsky, I.; Nicks, J.; Dunn, W.A., Jr.; Notterpek, L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J. Neurosci. 2010, 30, 11388–11397. [Google Scholar] [CrossRef] [Green Version]
- Nicks, J.; Lee, S.; Harris, A.; Falk, D.J.; Todd, A.G.; Arredondo, K.; Dunn, W.A., Jr.; Notterpek, L. Rapamycin improves peripheral nerve myelination while it fails to benefit neuromuscular performance in neuropathic mice. Neurobiol. Dis. 2014, 70, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Hantke, J.; Carty, L.; Wagstaff, L.J.; Turmaine, M.; Wilton, D.K.; Quintes, S.; Koltzenburg, M.; Baas, F.; Mirsky, R.; Jessen, K.R. c-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain 2014, 137, 2922–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiepura, A.J.; Kochański, A. Charcot-Marie-Tooth type 1A drug therapies: Role of adenylyl cyclase activity and G-protein coupled receptors in disease pathomechanism. Acta Neurobiol. Exp. 2018, 78, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Verrier, J.D.; Lau, P.; Hudson, L.; Murashov, A.K.; Renne, R.; Notterpek, L. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia 2009, 57, 1265–1279. [Google Scholar] [CrossRef] [Green Version]
- Bremer, J.; O’Connor, T.; Tiberi, C.; Rehrauer, H.; Weis, J.; Aguzzi, A. Ablation of Dicer from Murine Schwann Cells Increases Their Proliferation while Blocking Myelination. PLoS ONE 2010, 5, e12450. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.; Baumann, R.; Norrmén, C.; Somandin, C.; Miehe, M.; Jacob, C.; Lühmann, T.; Hall-Bozic, H.; Mantei, N.; Meijer, D.; et al. Dicer in Schwann Cells Is Required for Myelination and Axonal Integrity. J. Neurosci. 2010, 30, 6763–6775. [Google Scholar] [CrossRef] [Green Version]
- Yun, B.; Anderegg, A.; Menichella, D.; Wrabetz, L.; Feltri, M.L.; Awatramani, R. MicroRNA-deficient Schwann cells display congenital hypomyelination. J. Neurosci. 2010, 30, 7722–7728. [Google Scholar] [CrossRef] [Green Version]
- Verrier, J.D.; Semple-Rowland, S.; Madorsky, I.; Papin, J.E.; Notterpek, L. Reduction of Dicer impairs Schwann cell differentiation and myelination. J. Neurosci. Res. 2010, 88, 2558–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viader, A.; Chang, L.-W.; Fahrner, T.; Nagarajan, R.; Milbrandt, J. MicroRNAs Modulate Schwann Cell Response to Nerve Injury by Reinforcing Transcriptional Silencing of Dedifferentiation-Related Genes. J. Neurosci. 2011, 31, 17358–17369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokey, N.G.; Srinivasan, R.; Lopez-Anido, C.; Krueger, C.; Svaren, J. Developmental Regulation of MicroRNA Expression in Schwann Cells. Mol. Cell. Biol. 2012, 32, 558–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gökbuget, D.; Pereira, J.A.; Opitz, L.; Christe, D.; Kessler, T.; Marchais, A.; Suter, U. The miRNA biogenesis pathway prevents inappropriate expression of injury response genes in developing and adult Schwann cells. Glia 2018, 66, 2632–2644. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. Step aside CRISPR, RNA editing is taking off. Nat. Cell Biol. 2020, 578, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.I.; Pleasure, D. Whither Hope for Pharmacological Treatment of Charcot-Marie-Tooth Disease Type 1A? JAMA Neurol. 2013, 70, 969–971. [Google Scholar] [CrossRef]
- Sufit, R.L.; Ajroud-Driss, S.; Casey, P.; Kessler, J.A. Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 269–278. [Google Scholar] [CrossRef]
- Kessler, J.A.; Shaibani, A.; Sang, C.N.; Christiansen, M.; Kudrow, D.; Vinik, A.; Shin, N. Gene therapy for diabetic peripheral neuropathy: A randomized, placebo-controlled phase III study of VM202, a plasmid DNA encoding human hepatocyte growth factor. Clin. Transl. Sci. 2021. [Google Scholar] [CrossRef]
- Kim, J.S.; Hwang, H.Y.; Cho, K.R.; Park, E.-A.; Lee, W.; Paeng, J.C.; Lee, D.S.; Kim, H.-K.; Sohn, D.-W.; Kim, K.-B. Intramyocardial transfer of hepatocyte growth factor as an adjunct to CABG: Phase I clinical study. Gene Ther. 2013, 20, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Gordon, P.H.; Moore, D.H.; Miller, R.G.; Florence, J.M.; Verheijde, J.L.; Doorish, C.; Hilton, J.F.; Spitalny, G.M.; MacArthur, R.B.; Mitsumoto, H.; et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007, 6, 1045–1053. [Google Scholar] [CrossRef]
- Attarian, S.; Vallat, J.-M.; Magy, L.; Funalot, B.; Gonnaud, P.-M.; Lacour, A.; Péréon, Y.; Dubourg, O.; Pouget, J.; Micallef, J.; et al. An exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet J. Rare Dis. 2014, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Kaya, F.; Belin, S.; Diamantidis, G.; Fontes, M. Ascorbic acid is a regulator of the intracellular cAMP concentration: Old molecule, new functions? FEBS Lett. 2008, 582, 3614–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, M.J.; Lochmüller, A.; Atalaia, A.; Horvath, R. Targeted Therapies for Hereditary Peripheral Neuropathies: Systematic Review and Steps Towards a ’treatabolome. J. Neuromuscul. Dis. 2021, 8, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.-A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [Green Version]
- Khajavi, M.; Inoue, K.; Wiszniewski, W.; Ohyama, T.; Snipes, G.J.; Lupski, J.R. Curcumin Treatment Abrogates Endoplasmic Reticulum Retention and Aggregation-Induced Apoptosis Associated with Neuropathy-Causing Myelin Protein Zero–Truncating Mutants. Am. J. Hum. Genet. 2005, 77, 841–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillaud, M.; Msheik, Z.; Ndong-Ntoutoume, G.M.-A.; Vignaud, L.; Richard, L.; Favreau, F.; Faye, P.-A.; Sturtz, F.; Granet, R.; Vallat, J.-M.; et al. Curcumin–cyclodextrin/cellulose nanocrystals improve the phenotype of Charcot-Marie-Tooth-1A transgenic rats through the reduction of oxidative stress. Free. Radic. Biol. Med. 2020, 161, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Chahbouni, M.; López, M.D.S.; Molina-Carballo, A.; De Haro, T.; Muñoz-Hoyos, A.; Fernández-Ortiz, M.; Guerra-Librero, A.; Acuña-Castroviejo, D. Melatonin Treatment Reduces Oxidative Damage and Normalizes Plasma Pro-Inflammatory Cytokines in Patients Suffering from Charcot-Marie-Tooth Neuropathy: A Pilot Study in Three Children. Molecular 2017, 22, 1728. [Google Scholar] [CrossRef] [Green Version]
- Chahbouni, M.; Escames, G.; López, L.C.; Sevilla, B.; Doerrier, C.; Muñoz-Hoyos, A.; Molina-Carballo, A.; Acuña-Castroviejo, D. Melatonin treatment counteracts the hyperoxidative status in erythrocytes of patients suffering from Duchenne muscular dystrophy. Clin. Biochem. 2011, 44, 853–858. [Google Scholar] [CrossRef]
- Désarnaud, F.; Thi, A.N.D.; Brown, A.M.; Lemke, G.; Suter, U.; Baulieu, E.-E.; Schumacher, M. Progesterone Stimulates the Activity of the Promoters of Peripheral Myelin Protein-22 and Protein Zero Genes in Schwann Cells. J. Neurochem. 2002, 71, 1765–1768. [Google Scholar] [CrossRef]
- Melcangi, R.C.; Magnaghi, V.; Cavarretta, I.; Zucchi, I.; Bovolin, P.; D’Urso, D.; Martini, L. Progesterone derivatives are able to influence peripheral myelin protein 22 and P0 gene expression: Possible mechanisms of action. J. Neurosci. Res. 1999, 56, 349–357. [Google Scholar] [CrossRef]
- Robertson, A.M.; Huxley, C.; King, R.H.M.; Thomas, P.K. Development of early postnatal peripheral nerve abnormalities in Trembler-J and PMP22 transgenic mice. J. Anat. 1999, 195, 331–339. [Google Scholar] [CrossRef]
- Schulz, A.; Kyselyova, A.; Baader, S.L.; Jung, M.J.; Zoch, A.; Mautner, V.-F.; Hagel, C.; Morrison, H. Neuronal merlin influences ERBB2 receptor expression on Schwann cells through neuregulin 1 type III signalling. Brain 2014, 137, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visigalli, D.; Capodivento, G.; Basit, A.; Fernández, R.; Hamid, Z.; Pencová, B.; Gemelli, C.; Marubbi, D.; Pastorino, C.; Luoma, A.M.; et al. Exploiting Sphingo- and Glycerophospholipid Impairment to Select Effective Drugs and Biomarkers for CMT1A. Front. Neurol. 2020, 11, 903. [Google Scholar] [CrossRef]
- Inglese, J.; Dranchak, P.; Moran, J.J.; Jang, S.-W.; Srinivasan, R.; Santiago, Y.; Zhang, L.; Guha, R.; Martinez, N.; MacArthur, R.; et al. Genome Editing-Enabled HTS Assays Expand Drug Target Pathways for Charcot–Marie–Tooth Disease. ACS Chem. Biol. 2014, 9, 2594–2602. [Google Scholar] [CrossRef]
- Jang, S.-W.; Lopez-Anido, C.; MacArthur, R.; Svaren, J.; Inglese, J. Identification of Drug Modulators Targeting Gene-Dosage Disease CMT1A. ACS Chem. Biol. 2012, 7, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrahari, A.K. A Computational Approach to Identify a Potential Alternative Drug With Its Positive Impact Toward PMP22. J. Cell. Biochem. 2017, 118, 3730–3743. [Google Scholar] [CrossRef] [PubMed]
- Chittoor-Vinod, V.G.; Lee, S.; Judge, S.; Notterpek, L. Inducible HSP70 Is Critical in Preventing the Aggregation and Enhancing the Processing of PMP22. ASN Neuro 2015, 7, 1759091415569909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, S.; Madorsky, I.; Pileggi, J.G.; Kamal, A.; Notterpek, L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol. Dis. 2008, 32, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, L.; Doyle, J.P.; Hensley, P.; Colman, D.R.; Hendrickson, W.A. Crystal Structure of the Extracellular Domain from P0, the Major Structural Protein of Peripheral Nerve Myelin. Neuron 1996, 17, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Morena, J.; Gupta, A.; Hoyle, J.C. Charcot-Marie-Tooth: From Molecules to Therapy. Int. J. Mol. Sci. 2019, 20, 3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrabetz, L.; D’Antonio, M.; Pennuto, M.; Dati, G.; Tinelli, E.; Fratta, P.; Previtali, S.; Imperiale, D.; Zielasek, J.; Toyka, K.; et al. Different Intracellular Pathomechanisms Produce Diverse Myelin Protein Zero Neuropathies in Transgenic Mice. J. Neurosci. 2006, 26, 2358–2368. [Google Scholar] [CrossRef]
- Saporta, M.A.C.; Shy, B.R.; Patzko, A.; Bai, Y.; Pennuto, M.; Ferri, C.; Tinelli, E.; Saveri, P.; Kirschner, D.; Crowther, M.; et al. MpzR98C arrests Schwann cell development in a mouse model of early-onset Charcot–Marie–Tooth disease type 1B. Brain 2012, 135, 2032–2047. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.P.; Martini, R.; Lemke, G.; Soriano, P.; Schachner, M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell 1992, 71, 565–576. [Google Scholar] [CrossRef]
- Chen, Y.; Podojil, J.R.; Kunjamma, R.B.; Jones, J.; Weiner, M.; Lin, W.; Miller, S.D.; Popko, B. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 2019, 142, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Patzkó, Á.; Bai, Y.; Saporta, M.A.; Katona, I.; Wu, X.; Vizzuso, D.; Feltri, M.L.; Wang, S.; Dillon, L.M.; Kamholz, J.; et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain 2012, 135, 3551–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Patzkó, Á.; Shy, M.E. Unfolded protein response, treatment and CMT1B. Rare Dis. (Austin, Tex.) 2013, 1, e24049. [Google Scholar] [CrossRef] [Green Version]
- Shy, M.E.; Jáni, A.; Krajewski, K.; Grandis, M.; Lewis, R.A.; Li, J.; Shy, R.R.; Balsamo, J.; Lilien, J.; Garbern, J.Y.; et al. Phenotypic clustering in MPZ mutations. Brain 2004, 127, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Scapin, C.; Ferri, C.; Pettinato, E.; Zambroni, D.; Bianchi, F.; Del Carro, U.; Belin, S.; Caruso, D.; Mitro, N.; Pellegatta, M.; et al. Enhanced axonal neuregulin-1 type-III signaling ameliorates neurophysiology and hypomyelination in a Charcot-Marie-Tooth type 1B mouse model. Hum. Mol. Genet. 2019, 28, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Scapin, C.; Ferri, C.; Pettinato, E.; Bianchi, F.; Del Carro, U.; Feltri, M.L.; Kaufman, R.J.; Wrabetz, L.; D’Antonio, M. Phosphorylation of eIF2α promotes Schwann cell differentiation and myelination in CMT1B mice with activated UPR. J. Neurosci. 2020, 40, 8174–8187. [Google Scholar] [CrossRef]
- Groh, J.; Basu, R.; Stanley, E.R.; Martini, R. Cell-Surface and Secreted Isoforms of CSF-1 Exert Opposing Roles in Macrophage-Mediated Neural Damage in Cx32-Deficient Mice. J. Neurosci. 2016, 36, 1890–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawade, L.; Grandi, F.; Mignanelli, M.; Patiño-López, G.; Klinkert, K.; Langa-Vives, F.; Di Guardo, R.; Echard, A.; Bolino, A.; Haucke, V. Rab35-regulated lipid turnover by myotubularins represses mTORC1 activity and controls myelin growth. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Bolino, A.; Piguet, F.; Alberizzi, V.; Pellegatta, M.; Rivellini, C.; Guerrero-Valero, M.; Noseda, R.; Brombin, C.; Nonis, A.; D’Adamo, P.; et al. Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol. Med. 2016, 8, 1438–1454. [Google Scholar] [CrossRef]
- Presa, M.; Bailey, R.M.; Davis, C.; Murphy, T.; Cook, J.; Walls, R.; Wilpan, H.; Bogdanik, L.; Lenk, G.M.; Burgess, R.W.; et al. AAV9-mediated FIG4 delivery prolongs life span in Charcot Marie Tooth disease type 4J mouse model. J. Clin. Investig. 2021. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.O.; Bosanac, T.; Mao, X.; Engber, T.M.; DiAntonio, A.; Milbrandt, J.; Devraj, R.; Krauss, R. Small Molecule SARM1 Inhibitors Recapitulate the SARM1(-/-) Phenotype and Allow Recovery of a Metastable Pool of Axons Fated to Degenerate. Cell Rep. 2021, 34, 108588. [Google Scholar] [CrossRef]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, I.G.W. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 2020, 63, 7033–7051. [Google Scholar] [CrossRef] [PubMed]
- Hotta, N.; Akanuma, Y.; Kawamori, R.; Matsuoka, K.; Oka, Y.; Shichiri, M.; Toyota, T.; Nakashima, M.; Yoshimura, I.; Sakamoto, N.; et al. Long-Term Clinical Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy: The 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care 2006, 29, 1538–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, A.; Zhu, Y.; Rebelo, A.P.; Negri, S.; Courel, S.; Abreu, L.; Bacon, C.J.; Bai, Y.; Bis-Brewer, D.M.; Bugiardini, E.; et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020, 52, 473–481. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Kohara, N.; Baba, M.; Komori, T.; Naito, Y.; Imai, T.; Satoh, J.; Yamaguchi, Y.; Hamatani, T. Aldose reductase inhibitor ranirestat significantly improves nerve conduction velocity in diabetic polyneuropathy: A randomized double-blind placebo-controlled study in Japan. J. Diabetes Investig. 2018, 10, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Maciel, R.; Correa, R.; Taniguchi, J.B.; Araujo, I.P.; Saporta, M.A. Human Tridimensional Neuronal Cultures for Phenotypic Drug Screening in Inherited Peripheral Neuropathies. Clin. Pharmacol. Ther. 2019, 107, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Woo, S.-Y.; Hong, Y.B.; Choi, H.; Kim, J.; Choi, H.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with HSPB1 Mutation. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shababi, M.; Feng, Z.; Villalon, E.; Sibigtroth, C.M.; Osman, E.Y.; Miller, M.R.; Williams-Simon, P.A.; Lombardi, A.; Sass, T.H.; Atkinson, A.K.; et al. Rescue of a Mouse Model of Spinal Muscular Atrophy with Respiratory Distress Type 1 by AAV9-IGHMBP2 Is Dose Dependent. Mol. Ther. 2016, 24, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Clemens, M.J. Initiation Factor eIF2α Phosphorylation in Stress Responses and Apoptosis. Blue Biotechnol. 2001, 27, 57–89. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback Inhibition of the Unfolded Protein Response by GADD34-Mediated Dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- D’Antonio, M.; Musner, N.; Scapin, C.; Ungaro, D.; Del Carro, U.; Ron, D.; Feltri, M.L.; Wrabetz, L. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J. Exp. Med. 2013, 210, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Jessen, K.R.; Mirsky, R. Negative regulation of myelination: Relevance for development, injury, and demyelinating disease. Glia 2008, 56, 1552–1565. [Google Scholar] [CrossRef]
- Florio, F.; Ferri, C.; Scapin, C.; Feltri, M.L.; Wrabetz, L.; D’Antonio, M. Sustained Expression of Negative Regulators of Myelination Protects Schwann Cells from Dysmyelination in a Charcot–Marie–Tooth 1B Mouse Model. J. Neurosci. 2018, 38, 4275–4287. [Google Scholar] [CrossRef]
- Groh, J.; Weis, J.; Zieger, H.L.; Stanley, E.R.; Heuer, H.; Martini, R. Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot–Marie–Tooth disease type 1X. Brain 2011, 135, 88–104. [Google Scholar] [CrossRef] [Green Version]
- Klein, D.; Groh, J.; Weishaupt, A.; Martini, R. Endogenous antibodies contribute to macrophage-mediated demyelination in a mouse model for CMT1B. J. Neuroinflammation 2015, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D.; Patzkó, Á.; Schreiber, D.; Van Hauwermeiren, A.; Baier, M.; Groh, J.; West, B.L.; Martini, R. Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot–Marie–Tooth disease in mice. Brain 2015, 138, 3193–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleopa, K.A.; Abrams, C.K.; Scherer, S.S. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res. 2012, 1487, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleopa, K.A.; Scherer, S.S. Molecular genetics of X-linked Charcot-Marie-Tooth disease. Neuromolecular Med. 2006, 8, 107–122. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, F. A Review of X-linked Charcot-Marie-Tooth Disease. J. Child Neurol. 2015, 31, 761–772. [Google Scholar] [CrossRef]
- Scherer, S.S.; Deschênes, S.M.; Xu, Y.T.; Grinspan, J.B.; Fischbeck, K.H.; Paul, D.L. Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 1995, 15, 8281–8294. [Google Scholar] [CrossRef]
- Scherer, S.S.; Xu, Y.-T.; Messing, A.; Willecke, K.; Fischbeck, K.H.; Jeng, L.J.B. Transgenic Expression of Human Connexin32 in Myelinating Schwann Cells Prevents Demyelination in Connexin32-Null Mice. J. Neurosci. 2005, 25, 1550–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargiannidou, I.; Vavlitou, N.; Aristodemou, S.; Hadjisavvas, A.; Kyriacou, K.; Scherer, S.S.; Kleopa, K.A. Connexin32 Mutations Cause Loss of Function in Schwann Cells and Oligodendrocytes Leading to PNS and CNS Myelination Defects. J. Neurosci. 2009, 29, 4736–4749. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, P.J.; Rossor, A.M.; Horga, A.; Jaunmuktane, Z.; Carr, A.; Saveri, P.; Piscosquito, G.; Pareyson, D.; Laura, M.; Blake, J.C.; et al. Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurol. 2017, 88, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Omori, Y.; Mesnil, M.; Yamasaki, H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: Functional defects and dominant negative effects. Mol. Biol. Cell 1996, 7, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, T.; Satake, M.; Ohnishi, A.; Tsutsumi, Y.; Fujikura, Y. Mutations of connexin32 in charcot-marie-tooth disease type X interfere with cell-to-cell communication but not cell proliferation and myelin-specific gene expression. J. Neurosci. Res. 1998, 51, 154–161. [Google Scholar] [CrossRef]
- Kleopa, K.A.; Yum, S.W.; Scherer, S.S. Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J. Neurosci. Res. 2002, 68, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.W.; Kleopa, K.A.; Shumasb, S.; Scherer, S.S. Diverse Trafficking Abnormalities of Connexin32 Mutants Causing CMTX. Neurobiol. Dis. 2002, 11, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Deschênes, S.M.; Walcott, J.L.; Wexler, T.L.; Scherer, S.S.; Fischbeck, K.H. Altered Trafficking of Mutant Connexin32. J. Neurosci. 1997, 17, 9077–9084. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.E.M.; Mambetisaeva, E.T.; Archer, D.A.; George, C.H.; Evans, W.H. Analysis of gap junction assembly using mutated connexins detected in Charcot-Marie-Tooth X-linked disease. J. Neurochem. 2001, 74, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Ri, Y.; Bennett, M.V.; Trexler, E.; Verselis, V.K.; Bargiello, T.A. Changes in Permeability Caused by Connexin 32 Mutations Underlie X-Linked Charcot-Marie-Tooth Disease. Neuron 1997, 19, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Anzini, P.; Neuberg, D.H.-H.; Schachner, M.; Nelles, E.; Willecke, K.; Zielasek, J.; Toyka, K.V.; Suter, U.; Martini, R. Structural Abnormalities and Deficient Maintenance of Peripheral Nerve Myelin in Mice Lacking the Gap Junction Protein Connexin 32. J. Neurosci. 1997, 17, 4545–4551. [Google Scholar] [CrossRef]
- Kyriakoudi, S.; Sargiannidou, I.; Kagiava, A.; Olympiou, M.; Kleopa, K.A. Golgi-retained Cx32 mutants interfere with gene addition therapy for CMT1X. Hum. Mol. Genet. 2017, 26, 1622–1633. [Google Scholar] [CrossRef]
- Jeng, L.J.B.; Balice-Gordon, R.J.; Messing, A.; Fischbeck, K.H.; Scherer, S.S. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol. Cell. Neurosci. 2006, 32, 283–298. [Google Scholar] [CrossRef]
- Martini, R.; Willison, H.J. Neuroinflammation in the peripheral nerve: Cause, modulator, or bystander in peripheral neuropathies? Glia 2016, 64, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Groh, J.; Heinl, K.; Kohl, B.; Wessig, C.; Greeske, J.; Fischer, S.; Martini, R. Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot–Marie–Tooth 1X. Hum. Mol. Genet. 2010, 19, 3530–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, J.; Klein, I.; Hollmann, C.; Wettmarshausen, J.; Klein, D.; Martini, R. CSF-1-activated macrophages are target-directed and essential mediators of schwann cell dedifferentiation and dysfunction in Cx32-deficient mice. Glia 2015, 63, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Bolino, A.; Muglia, M.; Conforti, F.L.; LeGuern, E.; Salih, M.; Georgiou, D.-M.; Christodoulou, K.; Hausmanowa-Petrusewicz, I.; Mandich, P.; Schenone, A.; et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 2000, 25, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Azzedine, H.; Bolino, A.; Taïeb, T.; Birouk, N.; Di Duca, M.; Bouhouche, A.; Benamou, S.; Mrabet, A.; Hammadouche, T.; Chkili, T.; et al. Mutations in MTMR13, a New Pseudophosphatase Homologue of MTMR2 and Sbf1, in Two Families with an Autosomal Recessive Demyelinating Form of Charcot-Marie-Tooth Disease Associated with Early-Onset Glaucoma. Am. J. Hum. Genet. 2003, 72, 1141–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareyson, D.; Stojkovic, T.; Reilly, M.M.; Leonard-Louis, S.; Laurà, M.; Blake, J.; Parman, Y.; Battaloglu, E.; Tazir, M.; Bellatache, M.; et al. A multicenter retrospective study of charcot-marie-tooth disease type 4B (CMT4B) associated with mutations in myotubularin-related proteins (MTMRs). Ann. Neurol. 2019, 86, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Lukasova, M.; Hanson, J.; Tunaru, S.; Offermanns, S. Nicotinic acid (niacin): New lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol. Sci. 2011, 32, 700–707. [Google Scholar] [CrossRef]
- LeGuern, E.; Guilbot, A.; Kessali, M.; Ravisé, N.; Tassin, J.; Maisonobe, T.; Grid, D.; Brice, A. Homozygosity mapping of an autosomal recessive form of demyelinating Charcot-Marie-Tooth disease to chromosome 5q23-q33. Hum. Mol. Genet. 1996, 5, 1685–1688. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, E.; Zenker, J.; Charles, A.-S.D.P.; Stendel, C.; Roos, A.; Medard, J.-J.; Tricaud, N.; Kleine, H.; Luscher, B.; Weis, J.; et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 17528–17533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouttenoire, E.A.; Lupo, V.; Calpena, E.; Bartesaghi, L.; Schüpfer, F.; Médard, J.-J.; Maurer, F.; Beckmann, J.S.; Senderek, J.; Palau, F.; et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia 2013, 61, 1041–1051. [Google Scholar] [CrossRef]
- Cipriani, S.; Phan, V.; Médard, J.-J.; Horvath, R.; Lochmüller, H.; Chrast, R.; Roos, A.; Spendiff, S. Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C. Int. J. Mol. Sci. 2018, 19, 4072. [Google Scholar] [CrossRef] [Green Version]
- Stendel, C.; Roos, A.; Kleine, H.; Arnaud, E.; Özçelik, M.; Sidiropoulos, P.N.M.; Zenker, J.; Schüpfer, F.; Lehmann, U.; Sobota, R.M.; et al. SH3TC2, a protein mutant in Charcot–Marie–Tooth neuropathy, links peripheral nerve myelination to endosomal recycling. Brain 2010, 133, 2462–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, S.; Chiu, M.; Dacks, J.B.; Roberts, R.C. Exclusive expression of the Rab11 effector SH3TC2 in Schwann cells links integrin-α6 and myelin maintenance to Charcot-Marie-Tooth disease type 4C. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneja, M.; Burns, J.; Saporta, M.A.; Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 2019, 90, 58–67. [Google Scholar] [CrossRef]
- Ando, M.; Hashiguchi, A.; Okamoto, Y.; Yoshimura, A.; Hiramatsu, Y.; Yujiro, H.; Higuchi, Y.; Mitsui, J.; Ishiura, H.; Umemura, A.; et al. Clinical and genetic diversities of Charcot-Marie-Tooth disease withMFN2mutations in a large case study. J. Peripher. Nerv. Syst. 2017, 22, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations. J. Neurosci. 2007, 27, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Sci. 2015, 348, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Gerdts, J.; Summers, D.W.; Milbrandt, J.; DiAntonio, A. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 2016, 89, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, D.W.; DiAntonio, A.; Milbrandt, J. Mitochondrial Dysfunction Induces Sarm1-Dependent Cell Death in Sensory Neurons. J. Neurosci. 2014, 34, 9338–9350. [Google Scholar] [CrossRef] [Green Version]
- Summers, D.W.; Frey, E.; Walker, L.J.; Milbrandt, J.; DiAntonio, A. DLK Activation Synergizes with Mitochondrial Dysfunction to Downregulate Axon Survival Factors and Promote SARM1-Dependent Axon Degeneration. Mol. Neurobiol. 2020, 57, 1146–1158. [Google Scholar] [CrossRef]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martinez, A.; Whitworth, A.J.; Conforti, L.; et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 2020, 134, 104678. [Google Scholar] [CrossRef]
- Rojas, D.R.; Kuner, R.; Agarwal, N. Metabolomic signature of type 1 diabetes-induced sensory loss and nerve damage in diabetic neuropathy. J. Mol. Med. 2019, 97, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, P.J. Aldose Reductase, Still a Compelling Target for Diabetic Neuropathy. Curr. Drug Targets 2008, 9, 14–36. [Google Scholar] [CrossRef]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.-Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA Synthetase Mutations in Charcot-Marie-Tooth Disease Type 2D and Distal Spinal Muscular Atrophy Type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Motley, W.W.; Seburn, K.L.; Nawaz, M.H.; Miers, K.E.; Cheng, J.; Antonellis, A.; Green, E.D.; Talbot, K.; Yang, X.-L.; Fischbeck, K.H.; et al. Charcot-Marie-Tooth–Linked Mutant GARS Is Toxic to Peripheral Neurons Independent of Wild-Type GARS Levels. PLoS Genet. 2011, 7, e1002399. [Google Scholar] [CrossRef]
- De Jonghe, P.; Mersivanova, I.; Nelis, E.; Del Favero, J.; Martin, J.-J.; Van Broeckhoven, C.; Evgrafov, O.; Timmerman, V. Further evidence that neurofilament light chain gene mutations can cause Charcot-Marie-Tooth disease type 2E. Ann. Neurol. 2001, 49, 245–249. [Google Scholar] [CrossRef]
- Tradewell, M.L.; Durham, H.D.; Mushynski, W.E.; Gentil, B.J. Mitochondrial and Axonal Abnormalities Precede Disruption of the Neurofilament Network in a Model of Charcot-Marie-Tooth Disease Type 2E and Are Prevented by Heat Shock Proteins in a Mutant-Specific Fashion. J. Neuropathol. Exp. Neurol. 2009, 68, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Heilman, P.L.; Song, S.; Miranda, C.J.; Meyer, K.; Srivastava, A.; Knapp, A.; Wier, C.G.; Kaspar, B.K.; Kolb, S.J. HSPB1 mutations causing hereditary neuropathy in humans disrupt non-cell autonomous protection of motor neurons. Exp. Neurol. 2017, 297, 101–109. [Google Scholar] [CrossRef]
- Kalinski, A.L.; Kar, A.N.; Craver, J.; Tosolini, A.P.; Sleigh, J.N.; Lee, S.J.; Hawthorne, A.; Brito-Vargas, P.; Miller-Randolph, S.; Passino, R.; et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J. Cell Biol. 2019, 218, 1871–1890. [Google Scholar] [CrossRef] [Green Version]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Berghe, P.V.; Timmerman, V.; Robberecht, W.; Bosch, L.V.D. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1–induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Benoy, V.; Van Helleputte, L.; Prior, R.; D’Ydewalle, C.; Haeck, W.; Geens, N.; Scheveneels, W.; Schevenels, B.; Cader, Z.; Talbot, K.; et al. HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 2018, 141, 673–687. [Google Scholar] [CrossRef]
- Mo, Z.; Zhao, X.; Liu, H.; Hu, Q.; Chen, X.Q.; Pham, J.; Wei, N.; Liu, Z.; Zhou, J.; Burgess, R.W.; et al. Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy. Nat. Commun. 2018, 9, 1007. [Google Scholar] [CrossRef] [Green Version]
- Cottenie, E.; Kochanski, A.; Jordanova, A.; Bansagi, B.; Zimon, M.; Horga, A.; Jaunmuktane, Z.; Saveri, P.; Rasic, V.M.; Baets, J.; et al. Truncating and Missense Mutations in IGHMBP2 Cause Charcot-Marie Tooth Disease Type 2. Am. J. Hum. Genet. 2014, 95, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, K.; Schuelke, M.; Diers, A.; Hoffmann, K.; Lucke, B.; Adams, C.; Bertini, E.; Leonhardt-Horti, H.; Muntoni, F.; Ouvrier, R.; et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 2001, 29, 75–77. [Google Scholar] [CrossRef]
- Cox, G.A.; Mahaffey, C.L.; Frankel, W.N. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron 1998, 21, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Sadjadi, R.; Reilly, M.M.; Shy, M.E.; Pareyson, D.; Laura, M.; Murphy, S.; Feely, S.M.E.; Grider, T.; Bacon, C.; Piscosquito, G.; et al. Psychometrics evaluation of Charcot-Marie-Tooth Neuropathy Score (CMTNSv2) second version, using Rasch analysis. J. Peripher. Nerv. Syst. 2014, 19, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.M.; Herrmann, D.N.; McDermott, M.P.; Scherer, S.S.; Shy, M.E.; Reilly, M.M.; Pareyson, D. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Fridman, V.; Sillau, S.; Acsadi, G.; Bacon, C.; Dooley, K.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. A longitudinal study of CMT1A using Rasch analysis based CMT neuropathy and examination scores. Neurology 2020, 94, e884–e896. [Google Scholar] [CrossRef]
- Micallef, J.; Attarian, S.; Dubourg, O.; Gonnaud, P.-M.; Hogrel, J.-Y.; Stojkovic, T.; Bernard, R.; Jouve, E.; Pitel, S.; Vacherot, F.; et al. Effect of ascorbic acid in patients with Charcot–Marie–Tooth disease type 1A: A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2009, 8, 1103–1110. [Google Scholar] [CrossRef]
- Shy, M.E.; Chen, L.; Swan, E.R.; Taube, R.; Krajewski, K.M.; Herrmann, D.; Lewis, R.A.; McDermott, M.P. Neuropathy progression in Charcot-Marie-Tooth disease type 1A. Neurology 2008, 70, 378–383. [Google Scholar] [CrossRef]
- Burns, J.; Ouvrier, R.; Estilow, T.; Shy, R.; Laurá, M.; Pallant, J.F.; Lek, M.; Muntoni, F.; Reilly, M.M.; Pareyson, D.; et al. Validation of the Charcot-Marie-Tooth disease pediatric scale as an outcome measure of disability. Ann. Neurol. 2012, 71, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichinger, K.; Burns, J.; Cornett, K.; Bacon, C.; Shepherd, M.L.; Mountain, J.; Sowden, J.; Shy, R.; Shy, M.E.; Herrmann, D.N. The Charcot-Marie-Tooth Functional Outcome Measure (CMT-FOM). Neurology 2018, 91, e1381–e1384. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.; Cornett, K.M.D.; Estilow, T.; Pareyson, D.; Zuccarino, R.; Skorupinska, M.; Pipis, M.; Sowden, J.E.; Scherer, S.; Reilly, M.M.; et al. Reliability of the Charcot-Marie-Tooth functional outcome measure. J. Peripher. Nerv. Syst. 2020, 25, 288–291. [Google Scholar] [CrossRef]
- Bren, L. The importance of patient-reported outcomes. it’s all about the patients. FDA Consum. 2006, 40, 26–32. [Google Scholar]
- Johnson, N.E.; Heatwole, C.; Creigh, P.; McDermott, M.P.; Dilek, N.; Hung, M.; Bounsanga, J.; Tang, W.; Shy, M.E.; Herrmann, D.N. The Charcot-Marie-Tooth Health Index: Evaluation of a Patient-Reported Outcome. Ann. Neurol. 2018, 84, 225–233. [Google Scholar] [CrossRef]
- Menotti, F.; Laudani, L.; Damiani, A.; Macaluso, A. Amount and intensity of daily living activities in Charcot-Marie-Tooth 1A patients. Brain Behav. 2013, 4, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Padua, L.; Pazzaglia, C.; Pareyson, D.; Schenone, A.; Aiello, A.; Fabrizi, G.M.; Cavallaro, T.; Santoro, L.; Manganelli, F.; Gemignani, F.; et al. Novel outcome measures for Charcot-Marie-Tooth disease: Validation and reliability of the 6-min walk test and StepWatch(™) Activity Monitor and identification of the walking features related to higher quality of life. Eur. J. Neurol. 2016, 23, 1343–1350. [Google Scholar] [CrossRef]
- Morrow, J.M.; Sinclair, C.D.J.; Fischmann, A.; Reilly, M.M.; Hanna, M.G.; Yousry, T.A.; Thornton, J.S. Reproducibility, and age, body-weight and gender dependency of candidate skeletal muscle MRI outcome measures in healthy volunteers. Eur. Radiol. 2014, 24, 1610–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, J.M.; Sinclair, C.D.J.; Fischmann, A.; Machado, P.M.; Reilly, M.M.; Yousry, T.A.; Thornton, J.S.; Hanna, M.G. MRI biomarker assessment of neuromuscular disease progression: A prospective observational cohort study. Lancet Neurol. 2016, 15, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.M.; Evans, M.R.; Grider, T.; Sinclair, C.D.; Thedens, D.; Shah, S.; Yousry, T.A.; Hanna, M.G.; Nopoulos, P.; Thornton, J.S.; et al. Validation of MRC Centre MRI calf muscle fat fraction protocol as an outcome measure in CMT1A. Neurology 2018, 91, e1125–e1129. [Google Scholar] [CrossRef] [PubMed]
- Sandelius, Å.; Zetterberg, H.; Blennow, K.; Adiutori, R.; Malaspina, A.; Laura, M.; Reilly, M.M.; Rossor, A.M. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology 2018, 90, e518–e524. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Davison, M.; Wang, K.; Xia, T.; Kramer, M.; Call, K.; Luo, J.; Wu, X.; Zuccarino, R.; Bacon, C.; et al. Transmembrane protease serine 5: A novel Schwann cell plasma marker for CMT1A. Ann. Clin. Transl. Neurol. 2020, 7, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Shin, Y.K.; Jo, Y.R.; Park, D.K.; Song, M.-Y.; Yoon, B.-A.; Nam, S.-H.; Kim, J.H.; Choi, B.-O.; Shin, H.Y.; et al. p75 and neural cell adhesion molecule 1 can identify pathologic Schwann cells in peripheral neuropathies. Ann. Clin. Transl. Neurol. 2019, 6, 1292–1301. [Google Scholar] [CrossRef]
- Ko, H.-G.; Choi, J.-H.; Park, D.I.; Kang, S.J.; Lim, C.-S.; Sim, S.-E.; Shim, J.; Kim, J.-I.; Kim, S.; Choi, T.-H.; et al. Rapid Turnover of Cortical NCAM1 Regulates Synaptic Reorganization after Peripheral Nerve Injury. Cell Rep. 2018, 22, 748–759. [Google Scholar] [CrossRef] [Green Version]
- Rossor, A.; Shy, M.; Reilly, M. Are we prepared for clinical trials in Charcot-Marie-Tooth disease? Brain Res. 2020, 1729, 146625. [Google Scholar] [CrossRef] [PubMed]
- Pallmann, P.; Bedding, A.W.; Choodari-Oskooei, B.; Dimairo, M.; Flight, L.; Hampson, L.V.; Holmes, J.; Mander, A.P.; Odondi, L.; Sydes, M.R.; et al. Adaptive designs in clinical trials: Why use them, and how to run and report them. BMC Med. 2018, 16, 1–15. [Google Scholar] [CrossRef]


{kind=link}
| Compound | Rationale | Evaluation Stage | References |
|---|---|---|---|
| CLINICAL TRIALS | |||
| Gene Therapy | |||
| AAV1 delivered NT-3 | Neurotrophic activity | Phase I/II Clinical trial | [29,43] (NCT03520751) |
| Drug Therapy | |||
| PXT3003: cocktail of baclofen, naltrexone and sorbitol | Downregulation of PMP22 overexpression | Phase III Clinical trial | [63,64,65] (NCT04762758) |
| Melatonin | Anti-oxidant activity | Pilot clinical trial | [66] |
| Dietary lipid supplementation | Stimulates myelin biosynthesis | Pilot clinical trial | [67,68] |
| Ascorbic acid | Inhibition of cAMP pathway downregulates PMP22 overexpression | Phase II Clinical trial/Unsuccessful | [69,70,71,72,73] (NCT00484510) |
| Progesterone receptor antagonist (EllaOne®) | Anti-progesterone activity, inhibition of myelin-related genes expression in SCs | Phase II Clinical trial/Unsuccessful | (NCT02600286) |
| PRE-CLINICAL STUDIES | |||
| Gene Therapy | |||
| VM202: novel genomic HGF cDNA hybrid | Stimulation of SC repair and regeneration | [74,75] | |
| siRNA | Allele specific downregulation of PMP22 overexpression | [76] | |
| siRNA | Downregulation of P2RX7 overexpression to reduce abnormal Ca2+ influx into SC | [77] | |
| siRNA conjugated to squalenoyl nanoparticles | Downregulation of PMP22 overexpression | [44] | |
| shRNA | Downregulation of PMP22 overexpression | [42] | |
| Lentiviral delivered miR-318 | Overexpression of miR-318 downregulates overexpressed PMP22 | [26] | |
| AAV2 delivered miR-29a | Overexpression of miR-29a downregulates overexpressed PMP22 | [30] | |
| ASOs | Downregulation of PMP22 overexpression | [78] | |
| Antiparallel triplex-forming oligonucleotides | Bind on PMP22 promoters to downregulate overexpressed PMP22 | [79] | |
| CRISPR/Cas9 | Deletion of TATA-box of PMP22 gene promoter in order to downregulate PMP22 overexpression | [41] | |
| Drug Therapy | |||
| P2X7 Inhibitor | Downregulation of P2RX7 overexpression in order to reduce abnormal Ca2+ influx into SC | [77] | |
| CKD-504 | HDAC6 inhibitor, downregulation of PMP22 overexpression | [80] | |
| Nano-Cur | Modified curcumin, anti-oxidant activity | [81,82] | |
| Progesterone receptor antagonist (Onapristone) | Inhibition of SCs myelin-related genes expression | [83,84] | |
| NRG1 | Paracrine growth factor, genetic ablation | [85] | |
| Fasting and rapamycin | Improve ER processing of overproduced PMP22 | [86,87,88] | |
| Upregulation of c-Jun transcription factor | Stimulates myelin gene expression | [89,90] | |
| Compound | Rationale | References |
|---|---|---|
| Demyelinating CMT neuropathies | ||
| CMT1B | ||
| CLINICAL TRIALS | ||
| Gene Therapy | ||
| IFB-088: Sephin1 | Proteostasis restoring, clinical testing for safety | [129], (NCT03610334) |
| PRE-CLINICAL STUDIES | ||
| Drug Therapy | ||
| Curcumin | Anti-oxidant activity | [110,130,131,132] |
| NRG1-III | Activates transcriptional regulators of myelin genes | [133] |
| Genetic inactivation of Gadd34 | Upregulation of eIF2α phosphorylation controlling translation | [134] |
| CMT1X | ||
| Gene Therapy | ||
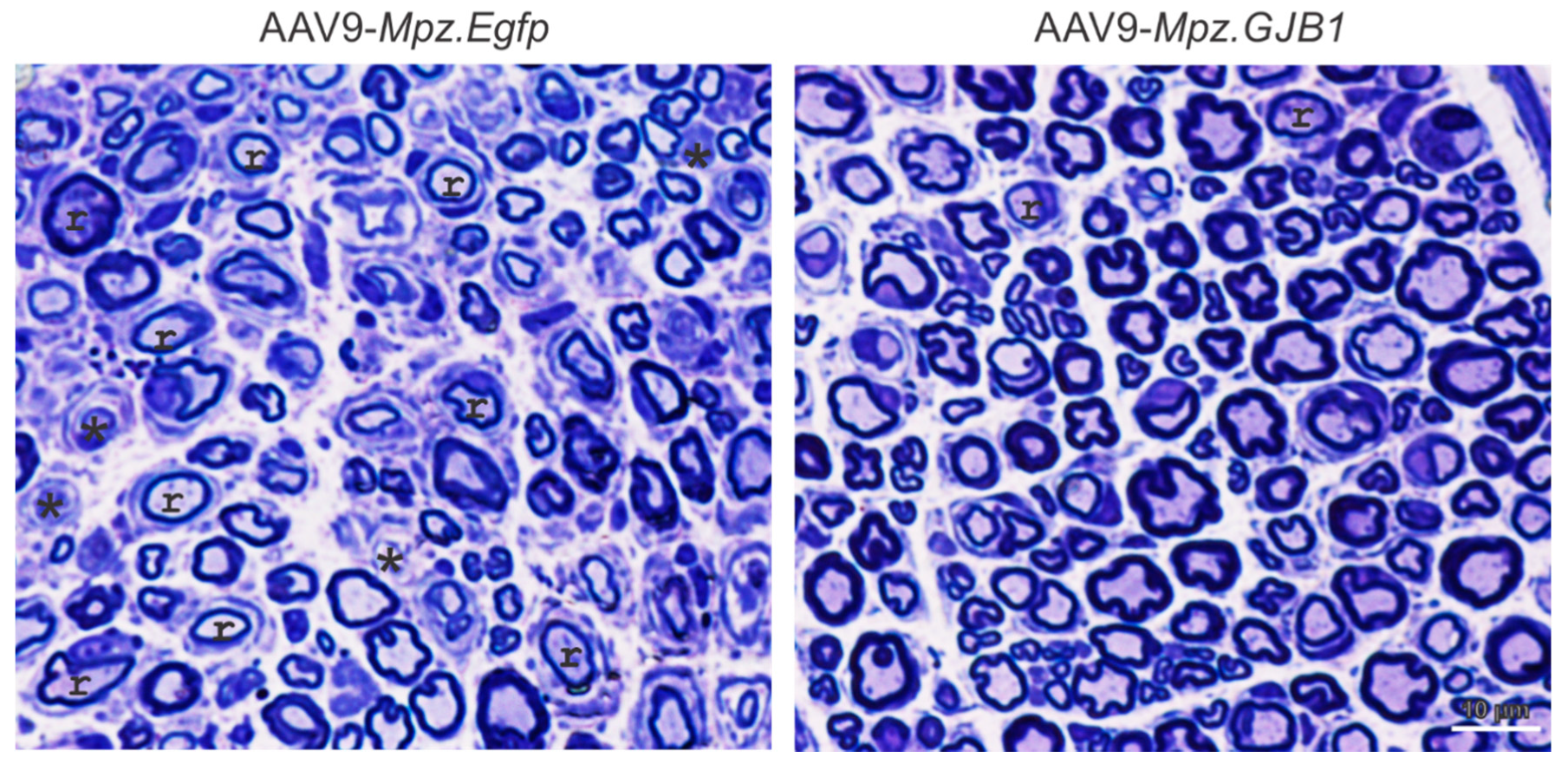
| Lentiviral delivered GJB1 gene | Schwann cell specific Cx32 production | [21,24] |
| AAV9 delivered GJB1 gene | Schwann cell specific Cx32 production | [34] |
| AAV1 delivered NT3 | Neurotrophic factor expression | [33] |
| Drug Therapy | ||
| CSF1 receptor antagonists | Blocking CSF-1 effects reduces inflammation in CMT1X model | [135] |
| CMT4B | ||
| Drug Therapy | ||
| Rapamycin | mTORC1 inhibitor restores Rab35 regulatory role on myelin production | [136] |
| Niaspan | Enhancing Tace activity to downregulate Nrg1 type III signaling | [137] |
| CMT4C | ||
| Gene Therapy | ||
| Lentiviral vector delivered SH3TC2 | Schwann cell specific SH3TC2 gene expression | [25] |
| CMT4J | ||
| Gene Therapy | ||
| AAV9 delivered FIG4 gene | Restoration of FIG4 expression | [138] |
| Axonal CMT neuropathies | ||
| CMT2A | ||
| Gene Therapy | ||
| AAV8 delivered SARM1 mutants | Dominant negative mutants block the wild type SARM1 function | [47] |
| MFN1 genetic addition | Compensates mutated MFN2 dysfunction | [139] |
| Drug Therapies | ||
| Isoquinoline | Inhibition the SARM1 NADase activity | [140] |
| MFN2 agonists | Improvement of mitochondrial trafficking | [141] |
| 6-phenylhexanamide derivative mitofusin activators | Improvement of mitochondrial motility | [142] |
| dHMN-SORD | ||
| Drug Therapies | ||
| Epalrestat | Aldose reductase inhibitor that normalizes abnormal sorbitol levels | [143,144] |
| Ranirestat | Aldose reductase inhibitor that normalizes abnormal sorbitol levels | [144,145] |
| CMT2D | ||
| Gene Therapy | ||
| AAV9-delivered artificialmiRNA | Allele-specific knockdown of dominant GARS mutants | [31] |
| CMT2E | ||
| Drug Therapies | ||
| Serine/threonine kinase inhibitors | Partially reverse neurofilament deposits phenotype in motor neuron axons | [146] |
| CMT2F | ||
| Drug Therapies | ||
| HDAC6 inhibitors | Restored acetylated α-tubulin levels improving mitochondrial mobility | [147] |
| CMT2S | ||
| Gene Therapy | ||
| AAV9-delivered IGHMBP2 gene | Restoration of IGHMBP2 gene function | [45,148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavrou, M.; Sargiannidou, I.; Georgiou, E.; Kagiava, A.; Kleopa, K.A. Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. Int. J. Mol. Sci. 2021, 22, 6048. https://doi.org/10.3390/ijms22116048
Stavrou M, Sargiannidou I, Georgiou E, Kagiava A, Kleopa KA. Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. International Journal of Molecular Sciences. 2021; 22(11):6048. https://doi.org/10.3390/ijms22116048
Chicago/Turabian StyleStavrou, Marina, Irene Sargiannidou, Elena Georgiou, Alexia Kagiava, and Kleopas A. Kleopa. 2021. "Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies" International Journal of Molecular Sciences 22, no. 11: 6048. https://doi.org/10.3390/ijms22116048
APA StyleStavrou, M., Sargiannidou, I., Georgiou, E., Kagiava, A., & Kleopa, K. A. (2021). Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. International Journal of Molecular Sciences, 22(11), 6048. https://doi.org/10.3390/ijms22116048