
The Influence of VE-Cadherin on Adhesion and Incorporation of Breast Cancer Cells into Vascular Endothelium

 , and
, and 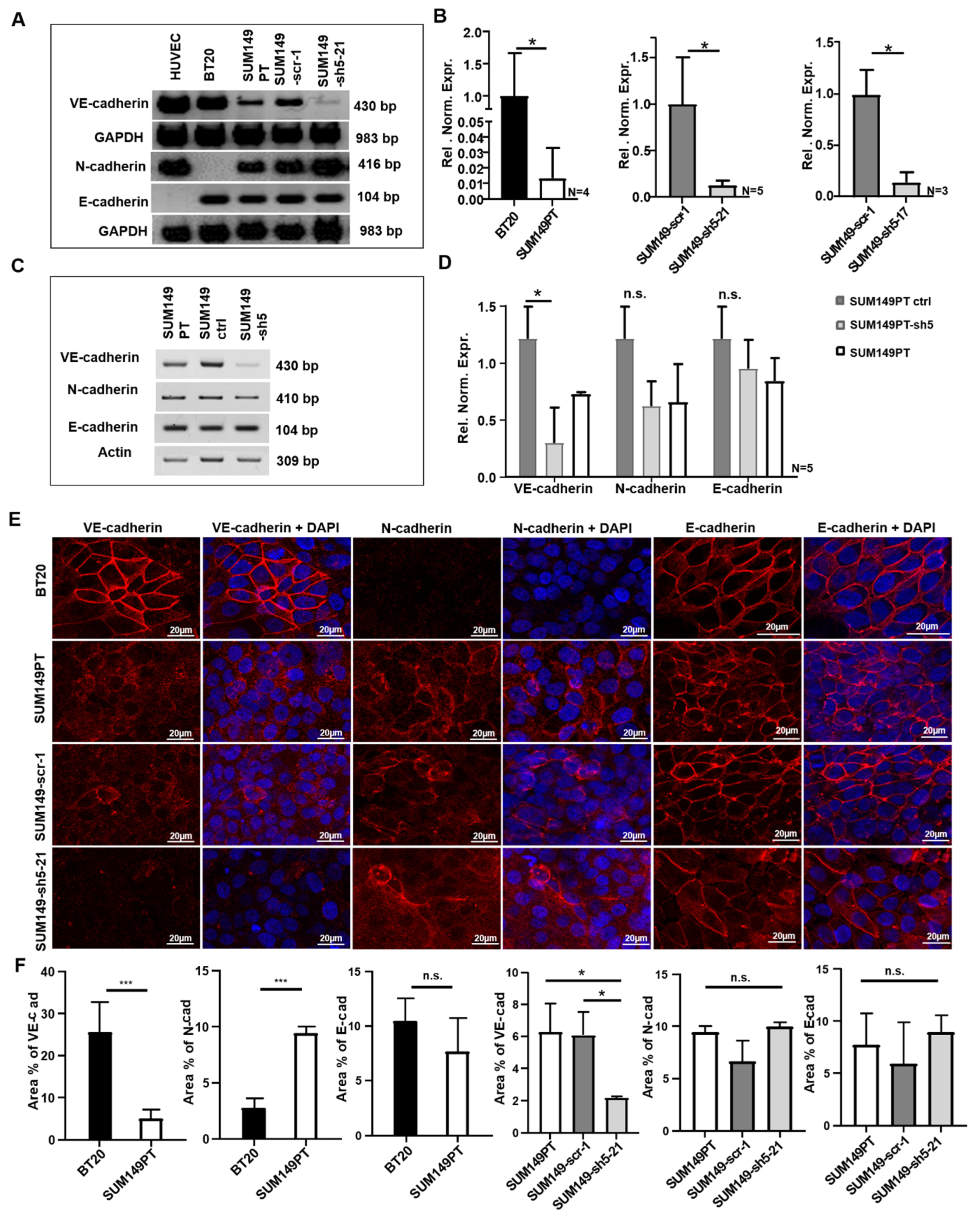{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. BT20 und SUM149 Cells Differentially Express VE-, N- and E-Cadherin
2.2. BT20, SUM149PT, SUM149-Scr-1 and SUM149-sh5-21 Cells Express VE-, E- and N-Cadherin on the Cell Surface
2.3. VE-Cadherin Enhances Breast Cancer Cell Adhesion to HUVEC Endothelium
2.4. BT20, SUM149PT, and VE-Cadherin Deficient SUM149PT Cells Incorporate into HUVEC Endothelium
2.5. SUM149PT Cells Incorporate More Frequently Than BT20 Cells into HUVEC Endothelium
2.6. VE-Cadherin Has no Quantitative Influence on the Incorporation Rate of Breast Cancer Cells
2.7. VE-Cadherin Has No Influence on Incorporation Inception Time or Duration
2.8. N-Cadherin Influences Adhesion and Incorporation of SUM149 Cells
2.9. BT20, SUM149PT, SUM149-Scr-1 and SUM149-sh5-21 Cells Interact With Endothelial VE-Cadherin during Incorporation
2.10. BT20, SUM149PT, SUM149-Scr-1 and SUM149-sh5-21 Cells Disrupt the Endothelial VE-Cadherin Barrier during Incorporation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of VE-Cadherin Knockdown Breast Cancer Cells
4.3. RNA Isolation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
4.4. Quantitative Reverse Transcription-PCR (qRT-PCR)
4.5. Immunofluorescence
4.6. Adhesion Assay
4.7. Incorporation Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Multitasking in tumor progression: Signaling functions of cell adhesion molecules. Ann. N. Y. Acad. Sci. 2004, 1014, 58–66. [Google Scholar] [CrossRef]
- van Sluis, G.L.; Niers, T.M.; Esmon, C.T.; Tigchelaar, W.; Richel, D.J.; Buller, H.R.; van Noorden, C.J.; Spek, C.A. Endogenous activated protein C limits cancer cell extravasation through sphingosine-1-phosphate receptor 1-mediated vascular endothelial barrier enhancement. Blood 2009, 114, 1968–1973. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Fraemohs, L.; Dejana, E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 2007, 7, 467–477. [Google Scholar] [CrossRef]
- Dejana, E. Endothelial cell–cell junctions: Happy together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Breier, G. Functions of the VEGF/VEGF Rezeptor System in the Vascular System. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2000; Volume 26, pp. 553–559. [Google Scholar]
- Reymond, N.; D’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous Expression of N-Cadherin in Breast Cancer Cells Induces Cell Migration, Invasion, and Metastasis. J. Cell Biol. 2000, 148, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial—Mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Schnittler, H.J.; Aust, D.E.; Friedrich, K.; Baretton, G.; Vestweber, D.; Breier, G. Vascular endothelial cadherin promotes breast cancer progression via transforming growth factor beta signaling. Cancer Res. 2008, 68, 1388–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, M.; Friedrich, K.; Wielockx, B.; Kuzmanov, A.; Kettelhake, A.; Labelle, M.; Schnittler, H.; Baretton, G.; Breier, G. Interplay between neural-cadherin and vascular endothelial-cadherin in breast cancer progression. Breast Cancer Res. 2012, 14, R154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breier, G.; Grosser, M.; Rezaei, M. Endothelial cadherins in cancer. Cell Tissue Res. 2014, 355, 523–527. [Google Scholar] [CrossRef]
- Fry, S.A.; Sinclair, J.; Timms, J.F.; Leathem, A.J.; Dwek, M.V. A targeted glycoproteomic approach identifies cadherin-5 as a novel biomarker of metastatic breast cancer. Cancer Lett. 2013, 328, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Cao, J.; Friedrich, K.; Kemper, B.; Brendel, O.; Grosser, M.; Adrian, M.; Baretton, G.; Breier, G.; Schnittler, H.-J. The expression of VE-cadherin in breast cancer cells modulates cell dynamics as a function of tumor differentiation and promotes tumor—Endothelial cell interactions. Histochem. Cell Biol. 2018, 149, 15–30. [Google Scholar] [CrossRef]
- Roh-Johnson, M.; Bravo-Cordero, J.J.; Patsialou, A.; Sharma, V.P.; Guo, P.; Liu, H.; Hodgson, L.; Condeelis, J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation HHS Public Access. Oncogene 2014, 33, 4203–4212. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Peterse, J.L.; Veer, L.J.V. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.M.; Munn, L.; Jain, R.K. Vasculogenic Mimicry: How Convincing, How Novel, and How Significant? Am. J. Pathol. 2000, 156, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Bartolomé, R.A.; Torres, S.; De Val, S.I.; Escudero-Paniagua, B.; Calviño, E.; Teixidó, J.; Casal, J.I. VE-cadherin RGD motifs promote metastasis and constitute a potential therapeutic target in melanoma and breast cancers. Oncotarget 2016, 8, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin Switch in Tumor Progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-Cadherin Promotes Motility in Human Breast Cancer Cells Regardless of Their E-Cadherin Expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.J.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.C.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef]
- Qi, J.; Chen, N.; Wang, J.; Siu, C.-H. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol. Biol. Cell 2005, 16, 4386–4397. [Google Scholar] [CrossRef] [PubMed]
- Hamilla, S.M.; Stroka, K.M.; Aranda-Espinoza, H. VE-Cadherin-Independent Cancer Cell Incorporation into the Vascular Endothelium Precedes Transmigration. PLoS ONE 2014, 9, e109748. [Google Scholar] [CrossRef]
- Fazakas, C.; Wilhelm, I.; Nagyőszi, P.; Farkas, A.E.; Haskó, J.; Molnár, J.; Bauer, H.; Bauer, H.C.; Ayaydin, F.; Dung, N.T.K.; et al. Transmigration of Melanoma Cells through the Blood-Brain Barrier: Role of Endothelial Tight Junctions and Melanoma-Released Serine Proteases. PLoS ONE 2011, 6, e20758. [Google Scholar] [CrossRef] [Green Version]
- Giebe, S.; Cockcroft, N.; Hewitt, K.; Brux, M.; Hofmann, A.; Morawietz, H.; Brunssen, C. Cigarette smoke extract counteracts atheroprotective effects of high laminar flow on endothelial function. Redox Biol. 2017, 12, 776–786. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real- Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 408, 402–408. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brock, T.; Boudriot, E.; Klawitter, A.; Großer, M.; Nguyen, T.T.P.; Giebe, S.; Klapproth, E.; Temme, A.; El-Armouche, A.; Breier, G. The Influence of VE-Cadherin on Adhesion and Incorporation of Breast Cancer Cells into Vascular Endothelium. Int. J. Mol. Sci. 2021, 22, 6049. https://doi.org/10.3390/ijms22116049
Brock T, Boudriot E, Klawitter A, Großer M, Nguyen TTP, Giebe S, Klapproth E, Temme A, El-Armouche A, Breier G. The Influence of VE-Cadherin on Adhesion and Incorporation of Breast Cancer Cells into Vascular Endothelium. International Journal of Molecular Sciences. 2021; 22(11):6049. https://doi.org/10.3390/ijms22116049
Chicago/Turabian StyleBrock, Thomas, Elisabeth Boudriot, Anke Klawitter, Marianne Großer, Trang T. P. Nguyen, Sindy Giebe, Erik Klapproth, Achim Temme, Ali El-Armouche, and Georg Breier. 2021. "The Influence of VE-Cadherin on Adhesion and Incorporation of Breast Cancer Cells into Vascular Endothelium" International Journal of Molecular Sciences 22, no. 11: 6049. https://doi.org/10.3390/ijms22116049
APA StyleBrock, T., Boudriot, E., Klawitter, A., Großer, M., Nguyen, T. T. P., Giebe, S., Klapproth, E., Temme, A., El-Armouche, A., & Breier, G. (2021). The Influence of VE-Cadherin on Adhesion and Incorporation of Breast Cancer Cells into Vascular Endothelium. International Journal of Molecular Sciences, 22(11), 6049. https://doi.org/10.3390/ijms22116049