
Migration and Adhesion of B-Lymphocytes to Specific Microenvironments in Mantle Cell Lymphoma: Interplay between Signaling Pathways and the Epigenetic Landscape


Abstract
:1. Introduction
2. Epigenetic Alteration in MCL
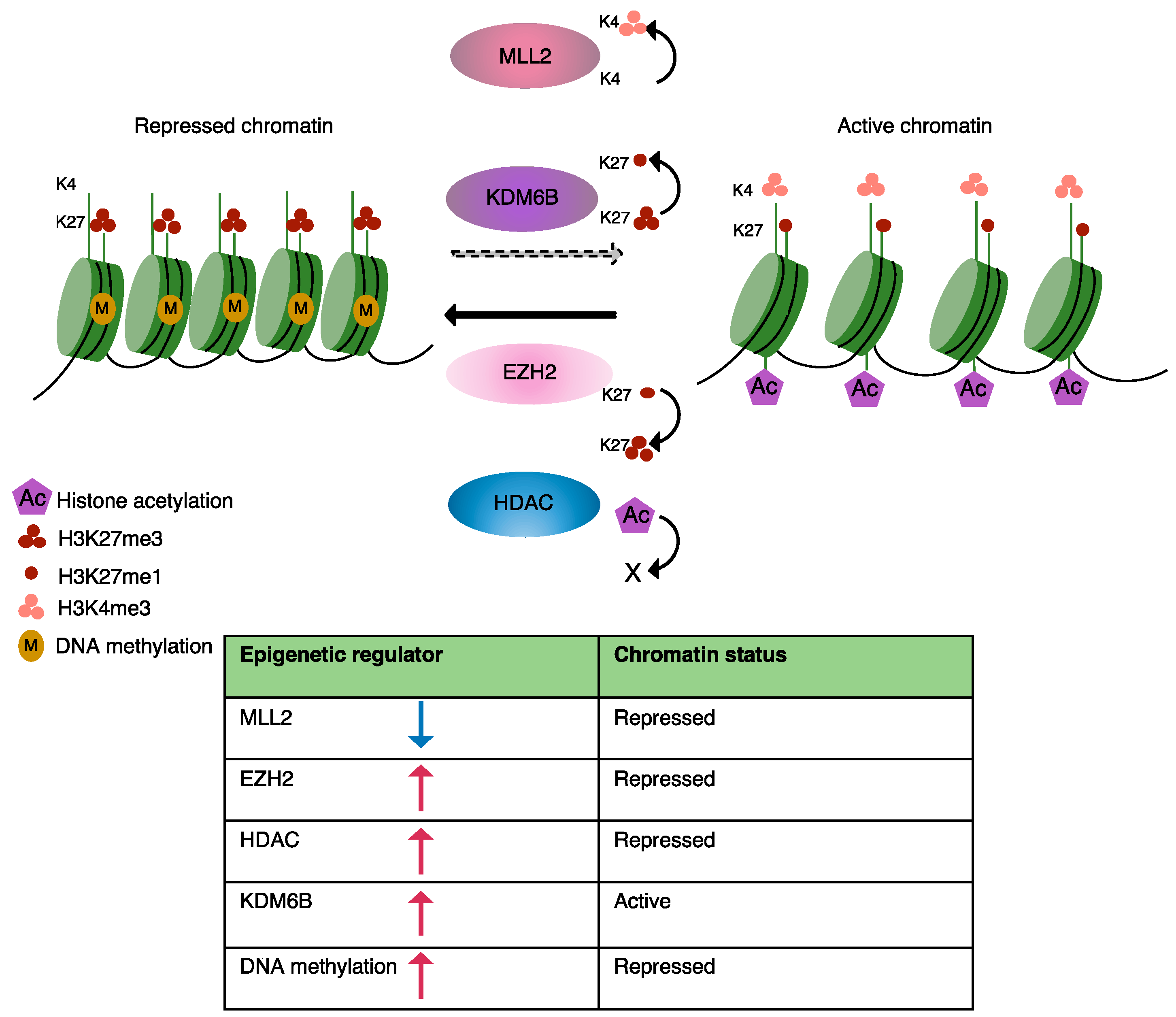
2.1. DNA Methylation
2.2. Chromatin Remodeling
2.3. Histone Deacetylation
2.4. Histone Methylation
2.5. Histone Demethylation
3. Tumor-Microenvironment Mediated Epigenetic Alterations
3.1. Role of the PI3K/AKT Pathway in Chromatin Modulation
3.2. NF-κB and Chromatin Remodeling
4. Targeting PI3K in MCL
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sadeghi, L.; Arvidsson, G.; Merrien, M.; Wasik, A.M.; Görgens, A.; Smith, C.E.; Sander, B.; Wright, A.P. Differential B-Cell Receptor Signaling Requirement for Adhesion of Mantle Cell Lymphoma Cells to Stromal Cells. Cancers 2020, 12, 1143. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Sonugur, F.G.; Akbulut, H. The Role of Tumor Microenvironment in Genomic Instability of Malignant Tumors. Front. Genet 2019, 10, 1063. [Google Scholar] [CrossRef]
- Arvidsson, G.; Henriksson, J.; Sander, B.; Wright, A.P. Mixed-species RNAseq analysis of human lymphoma cells adhering to mouse stromal cells identifies a core gene set that is also differentially expressed in the lymph node microenvironment of mantle cell lymphoma and chronic lymphocytic leukemia patients. Haematologica 2018, 103, 666–678. [Google Scholar] [CrossRef] [Green Version]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Ghielmini, M.; Zucca, E. How I treat mantle cell lymphoma. Blood 2009, 114, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Bosch, F.; Jares, P.; Campo, E.; Lopez-Guillermo, A.; Piris, M.A.; Villamor, N.; Tassies, D.; Jaffe, E.S.; Montserrat, E.; Rozman, C.; et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: A highly specific marker of mantle cell lymphoma. Blood 1994, 84, 2726–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffeld, M.; Jaffe, E.S. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood 1991, 78, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.Y.; Seymour, J.F.; Wang, M.L. Mantle Cell Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Edlefsen, K.L.; Greisman, H.A.; Yi, H.S.; Mantei, K.M.; Fromm, J.R. Early lymph node involvement by mantle cell lymphoma limited to the germinal center: Report of a case with a novel “follicular in situ” growth pattern. Am. J. Clin. Pathol. 2011, 136, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozos, A.; Royo, C.; Hartmann, E.; De Jong, D.; Baro, C.; Valera, A.; Fu, K.; Weisenburger, D.D.; Delabie, J.; Chuang, S.S.; et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009, 94, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
- Lord, M.; Wasik, A.M.; Christensson, B.; Sander, B. The utility of mRNA analysis in defining SOX11 expression levels in mantle cell lymphoma and reactive lymph nodes. Haematologica 2015, 100, e369–e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasik, A.M.; Priebe, V.; Lord, M.; Jeppsson-Ahlberg, A.; Christensson, B.; Sander, B. Flow cytometric analysis of SOX11: A new diagnostic method for distinguishing B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma from mantle cell lymphoma. Leuk. Lymphoma 2015, 56, 1425–1431. [Google Scholar] [CrossRef]
- Narurkar, R.; Alkayem, M.; Liu, D. SOX11 is a biomarker for cyclin D1-negative mantle cell lymphoma. Biomark Res. 2016, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bea, S.; Ribas, M.; Hernandez, J.M.; Bosch, F.; Pinyol, M.; Hernandez, L.; Garcia, J.L.; Flores, T.; Gonzalez, M.; Lopez-Guillermo, A.; et al. Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood 1999, 93, 4365–4374. [Google Scholar]
- Greiner, T.C.; Dasgupta, C.; Ho, V.V.; Weisenburger, D.D.; Smith, L.M.; Lynch, J.C.; Vose, J.M.; Fu, K.; Armitage, J.O.; Braziel, R.M.; et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2006, 103, 2352–2357. [Google Scholar] [CrossRef] [Green Version]
- Meissner, B.; Kridel, R.; Lim, R.S.; Rogic, S.; Tse, K.; Scott, D.W.; Moore, R.; Mungall, A.J.; Marra, M.A.; Connors, J.M.; et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood 2013, 121, 3161–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunawardana, J.; Jenkins, C.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, A.; Bigoni, R.; Rigolin, G.M.; Roberti, M.G.; Milani, R.; Bardi, A.; Minotto, C.; Agostini, P.; De Angeli, C.; Narducci, M.G.; et al. Acquired chromosome 11q deletion involving the ataxia teleangiectasia locus in B-cell non-Hodgkin’s lymphoma: Correlation with clinicobiologic features. J. Clin. Oncol. 2000, 18, 2607–2614. [Google Scholar] [CrossRef] [PubMed]
- Gumy-Pause, F.; Wacker, P.; Sappino, A.P. ATM gene and lymphoid malignancies. Leukemia 2004, 18, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, H.A.; Qi, X.; Jain, P.; Nomie, K.; Wang, Y.; Zhou, S.; Wang, M.L. Genetic mutations and features of mantle cell lymphoma: A systematic review and meta-analysis. Blood Adv. 2020, 4, 2927–2938. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; de Miranda, N.F.; Chen, L.; Wasik, A.M.; Mansouri, L.; Jurczak, W.; Galazka, K.; Dlugosz-Danecka, M.; Machaczka, M.; Zhang, H.; et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget 2016, 7, 38180–38190. [Google Scholar] [CrossRef]
- Di Pietro, A.; Good-Jacobson, K.L. Disrupting the Code: Epigenetic Dysregulation of Lymphocyte Function during Infectious Disease and Lymphoma Development. J. Immunol. 2018, 201, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Yusufova, N.; Kloetgen, A.; Teater, M.; Osunsade, A.; Camarillo, J.M.; Chin, C.R.; Doane, A.S.; Venters, B.J.; Portillo-Ledesma, S.; Conway, J.; et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 2021, 589, 299–305. [Google Scholar] [CrossRef]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Shaknovich, R.; Melnick, A. Epigenetics and B-cell lymphoma. Curr. Opin. Hematol. 2011, 18, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Enjuanes, A.; Albero, R.; Clot, G.; Navarro, A.; Bea, S.; Pinyol, M.; Martin-Subero, J.I.; Klapper, W.; Staudt, L.M.; Jaffe, E.S.; et al. Genome-wide methylation analyses identify a subset of mantle cell lymphoma with a high number of methylated CpGs and aggressive clinicopathological features. Int. J. Cancer 2013, 133, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmeray, E.; Kucuk, C. Genetic alterations in B cell lymphoma subtypes as potential biomarkers for noninvasive diagnosis, prognosis, therapy, and disease monitoring. Turk. J. Biol. 2020, 44, 1–14. [Google Scholar] [CrossRef]
- Wasik, A.M.; Wu, C.; Mansouri, L.; Rosenquist, R.; Pan-Hammarstrom, Q.; Sander, B. Clinical and functional impact of recurrent S1PR1 mutations in mantle cell lymphoma. Blood Adv. 2018, 2, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, J.; Qian, J.; Li, H.; Romaguera, J.E.; Kwak, L.W.; Wang, M.; Yi, Q. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood 2012, 120, 3783–3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, K.H.; Dalton, W.S.; Tao, J. The tumor microenvironment shapes hallmarks of mature B-cell malignancies. Oncogene 2015, 34, 4673–4682. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Fowler, N.H.; Cheah, C.Y.; Gascoyne, R.D.; Gribben, J.; Neelapu, S.S.; Ghia, P.; Bollard, C.; Ansell, S.; Curran, M.; Wilson, W.H.; et al. Role of the tumor microenvironment in mature B-cell lymphoid malignancies. Haematologica 2016, 101, 531–540. [Google Scholar] [CrossRef]
- Puente, X.S.; Jares, P.; Campo, E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Annese, T.; Ingravallo, G.; Tamma, R.; De Giorgis, M.; Maiorano, E.; Perrone, T.; Albano, F.; Specchia, G.; Ribatti, D. Inflammatory Infiltrate and Angiogenesis in Mantle Cell Lymphoma. Transl. Oncol. 2020, 13, 100744. [Google Scholar] [CrossRef]
- Rizzatti, E.G.; Falcao, R.P.; Panepucci, R.A.; Proto-Siqueira, R.; Anselmo-Lima, W.T.; Okamoto, O.K.; Zago, M.A. Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFbeta signalling pathways. Br. J. Haematol. 2005, 130, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Rassidakis, G.Z.; Medeiros, L.J.; Leventaki, V.; Keating, M.; McDonnell, T.J. Expression of STAT3 and its phosphorylated forms in mantle cell lymphoma cell lines and tumours. J. Pathol. 2003, 199, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.V.; Tamayo, A.T.; Yoshimura, L.C.; Lo, P.; Ford, R.J. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J. Immunol. 2003, 171, 88–95. [Google Scholar] [CrossRef]
- Kennedy, R.; Klein, U. Aberrant Activation of NF-kappaB Signalling in Aggressive Lymphoid Malignancies. Cells 2018, 7, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudelius, M.; Pittaluga, S.; Nishizuka, S.; Pham, T.H.; Fend, F.; Jaffe, E.S.; Quintanilla-Martinez, L.; Raffeld, M. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006, 108, 1668–1676. [Google Scholar] [CrossRef] [Green Version]
- Armengol, M.; Santos, J.C.; Fernandez-Serrano, M.; Profitos-Peleja, N.; Ribeiro, M.L.; Roue, G. Immune-Checkpoint Inhibitors in B-Cell Lymphoma. Cancers 2021, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Harrington, B.K.; Wheeler, E.; Hornbuckle, K.; Shana’ah, A.Y.; Youssef, Y.; Smith, L.; Hassan, Q., 2nd; Klamer, B.; Zhang, X.; Long, M.; et al. Modulation of immune checkpoint molecule expression in mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2498–2507. [Google Scholar] [CrossRef]
- Castillo, R.; Mascarenhas, J.; Telford, W.; Chadburn, A.; Friedman, S.M.; Schattner, E.J. Proliferative response of mantle cell lymphoma cells stimulated by CD40 ligation and IL-4. Leukemia 2000, 14, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusa, D.; Serra, S.; Coscia, M.; Rossi, D.; D’Arena, G.; Laurenti, L.; Jaksic, O.; Fedele, G.; Inghirami, G.; Gaidano, G.; et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica 2013, 98, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Qian, J.; Lu, Y.; Li, H.; Bao, H.; He, D.; Liu, Z.; Zheng, Y.; He, J.; Li, Y.; et al. Immune evasion of mantle cell lymphoma: Expression of B7-H1 leads to inhibited T-cell response to and killing of tumor cells. Haematologica 2013, 98, 1458–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Bai, C.; Gao, W.; Strom, T.B.; Rothstein, T.L. Suppression of expression and function of negative immune regulator PD-1 by certain pattern recognition and cytokine receptor signals associated with immune system danger. Int. Immunol. 2004, 16, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharawat, S.K. Epigenetic regulators of programmed death-ligand 1 expression in human cancers. Transl. Res. 2018, 202, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor kappa B pathways. Biomarker Res. 2012, 97, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, N.S.; Liu, D.; Herman, S.E.; Underbayev, C.; Tian, X.; Behrend, D.; Weniger, M.A.; Skarzynski, M.; Gyamfi, J.; Fontan, L.; et al. Pathogenic role of B-cell receptor signaling and canonical NF-kappaB activation in mantle cell lymphoma. Blood 2016, 128, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.Y.; Tian, C.; Zhang, Y.Z. [Research Advances in the Epigenetics of Diffuse Large B-Cell Lymphoma-Review]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2017, 25, 1232–1236. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Argatoff, L.H.; Connors, J.M.; Klasa, R.J.; Horsman, D.E.; Gascoyne, R.D. Mantle cell lymphoma: A clinicopathologic study of 80 cases. Blood 1997, 89, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Queiros, A.C.; Beekman, R.; Vilarrasa-Blasi, R.; Duran-Ferrer, M.; Clot, G.; Merkel, A.; Raineri, E.; Russinol, N.; Castellano, G.; Bea, S.; et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell 2016, 30, 806–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Li, Y.; Zhang, L.; Liu, X.; Feng, L.; Wang, X. The antitumor effects of arsenic trioxide in mantle cell lymphoma via targeting Wnt/betacatenin pathway and DNA methyltransferase-1. Oncol. Rep. 2017, 38, 3114–3120. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, T.; von Neuhoff, N.; Kamphues, K.; Emura, M.; Lehmann, U.; Tauscher, M.; Schraders, M.; Groenen, P.; Skawran, B.; Rudolph, C.; et al. Promoter methylation of PARG1, a novel candidate tumor suppressor gene in mantle-cell lymphomas. Haematologica 2007, 92, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jima, D.; Moffitt, A.B.; Liu, Q.; Czader, M.; Hsi, E.D.; Fedoriw, Y.; Dunphy, C.H.; Richards, K.L.; Gill, J.I.; et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014, 123, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Chan, Y.C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2019, 25, 119–129. [Google Scholar] [CrossRef]
- Xargay-Torrent, S.; Lopez-Guerra, M.; Saborit-Villarroya, I.; Rosich, L.; Campo, E.; Roue, G.; Colomer, D. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin. Cancer Res. 2011, 17, 3956–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, N.K.; Hatch, N.D.; Sutton, G.L.; Kling, M.; Vose, J.M.; Joshi, S.S. A novel approach to eliminate therapy-resistant mantle cell lymphoma: Synergistic effects of Vorinostat with Palbociclib. Leuk. Lymphoma 2019, 60, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, S.; Balasubramanian, S.; David, K.; Sirisawad, M.; Buggy, J.; Mauro, L.; Prachand, S.; Miller, R.; Gordon, L.I.; Evens, A.M. PCI-24781 Induces Caspase and Reactive Oxygen Species-Dependent Apoptosis Through NF-kappa B Mechanisms and Is Synergistic with Bortezomib in Lymphoma Cells. Clin. Cancer Res. 2009, 15, 3354–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Zeng, D.; Zhang, H.; Bell, T.; Yao, J.; Liu, Y.; Huang, S.; Li, C.J.; Lorence, E.; Zhou, S.; et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene 2019, 38, 1802–1814. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, W.; Wang, J.; Liu, Y.; An, R.; Jing, H. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018, 25, 129–140. [Google Scholar] [CrossRef]
- Oyer, J.A.; Huang, X.; Zheng, Y.; Shim, J.; Ezponda, T.; Carpenter, Z.; Allegretta, M.; Okot-Kotber, C.I.; Patel, J.P.; Melnick, A.; et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2014, 28, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L. The genetic basis of diffuse large B-cell lymphoma. Curr. Opin. Hematol. 2013, 20, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dominguez-Sola, D.; Hussein, S.; Lee, J.E.; Holmes, A.B.; Bansal, M.; Vlasevska, S.; Mo, T.; Tang, H.; Basso, K.; et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 2015, 21, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Kantidakis, T.; Saponaro, M.; Mitter, R.; Horswell, S.; Kranz, A.; Boeing, S.; Aygun, O.; Kelly, G.P.; Matthews, N.; Stewart, A.; et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016, 30, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demosthenous, C.; Gupta, S.K.; Sun, J.; Wang, Y.S.; Troska, T.P.; Gupta, M. Deregulation of Polycomb Repressive Complex-2 in Mantle Cell Lymphoma Confers Growth Advantage by Epigenetic Suppression ofcdkn2b. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, M.; Sander, B.; Ntoufa, S.; Papakonstantinou, N.; Sutton, L.A.; Stamatopoulos, K.; Kanduri, C.; Rosenquist, R. A key role for EZH2 in epigenetic silencing of HOX genes in mantle cell lymphoma. Epigenetics 2013, 8, 1280–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, R.; Sehgal, L.; Havranek, O.; Kohrer, S.; Khashab, T.; Jain, N.; Burger, J.A.; Neelapu, S.S.; Davis, R.E.; Samaniego, F. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica 2017, 102, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Ohguchi, H.; Harada, T.; Sagawa, M.; Kikuchi, S.; Tai, Y.T.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 2017, 31, 2661–2669. [Google Scholar] [CrossRef] [Green Version]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Varga-Weisz, P. ATP-dependent chromatin remodeling factors: Nucleosome shufflers with many missions. Oncogene 2001, 20, 3076–3085. [Google Scholar] [CrossRef] [Green Version]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.C.; Sethy, B.; Liou, J.P. Recent Update of HDAC Inhibitors in Lymphoma. Front. Cell Dev. Biol. 2020, 8, 576391. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Villagra, A.; Merino, O.; Rock-Klotz, J.; Woan, K.; Seto, E.; Martin, P.; Leonard, J.P.; Chen-Kiang, S.; Tao, J.G.; et al. HDAC Profiling In Mantle Cell Lymphoma Unveils HDAC11 and HDAC10 as Potential Molecular Targets. Blood 2010, 116, 1038–1039. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watters, J.M.; Wright, G.; Smith, M.A.; Shah, B.; Wright, K.L. Histone deacetylase 8 inhibition suppresses mantle cell lymphoma viability while preserving natural killer cell function. Biochem. Biophys. Res. Commun. 2021, 534, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Hu, Q.; Zhang, H.; Yang, F.; Peng, C.; Huang, C. HDAC3 Inhibition Upregulates PD-L1 Expression in B-Cell Lymphomas and Augments the Efficacy of Anti-PD-L1 Therapy. Mol. Cancer Ther. 2019, 18, 900–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharaj, K.; Powers, J.J.; Mediavilla-Varela, M.; Achille, A.; Gamal, W.; Quayle, S.; Jones, S.S.; Sahakian, E.; Pinilla-Ibarz, J. HDAC6 Inhibition Alleviates CLL-Induced T-Cell Dysfunction and Enhances Immune Checkpoint Blockade Efficacy in the Emu-TCL1 Model. Front. Immunol. 2020, 11, 590072. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, G.R.; Wang, Y.; Lu, T. Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 2011, 21, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.J.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear arnine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Kaniskan, H.U.; Jin, J. Recent progress in developing selective inhibitors of protein methyltransferases. Curr. Opin. Chem. Biol. 2017, 39, 100–108. [Google Scholar] [CrossRef]
- Schafer, V.; Ernst, J.; Rinke, J.; Winkelmann, N.; Beck, J.F.; Hochhaus, A.; Gruhn, B.; Ernst, T. EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 2016, 142, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Lesch, B.J. H3K4me1 Distribution Predicts Transcription State and Poising at Promoters. Front. Cell Dev. Biol. 2020, 8, 289. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Lachner, M.; O’Sullivan, R.J.; Jenuwein, T. An epigenetic road map for histone lysine methylation. J. Cell Sci. 2003, 116, 2117–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.H.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhart, S.H.; Kretzmer, H.; Holdt, L.M.; Juhling, F.; Ammerpohl, O.; Bergmann, A.K.; Northoff, B.H.; Doose, G.; Siebert, R.; Stadler, P.F.; et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016, 6, 37393. [Google Scholar] [CrossRef]
- Yin, S.; Yang, J.; Lin, B.; Deng, W.; Zhang, Y.; Yi, X.; Shi, Y.; Tao, Y.; Cai, J.; Wu, C.I.; et al. Exome sequencing identifies frequent mutation of MLL2 in non-small cell lung carcinoma from Chinese patients. Sci. Rep. 2014, 4, 6036. [Google Scholar] [CrossRef] [Green Version]
- Schuettengruber, B.; Cavalli, G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 2009, 136, 3531–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Kirmizis, A.; Bartley, S.M.; Kuzmichev, A.; Margueron, R.; Reinberg, D.; Green, R.; Farnham, P.J. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004, 18, 1592–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Su, I.H.; Basavaraj, A.; Krutchinsky, A.N.; Hobert, O.; Ullrich, A.; Chait, B.T.; Tarakhovsky, A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 2003, 4, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Velichutina, I.; Shaknovich, R.; Geng, H.; Johnson, N.A.; Gascoyne, R.D.; Melnick, A.M.; Elemento, O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 2010, 116, 5247–5255. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.W.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Li, B.; Chng, W.J. EZH2 abnormalities in lymphoid malignancies: Underlying mechanisms and therapeutic implications. J. Hematol. Oncol. 2019, 12, 118. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Cloos, P.A.C.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Gene Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.M.; Velusamy, T.; Chung, F.; Schaller, M.; Bailey, N.G.; Betz, B.L.; Miranda, R.N.; Porcu, P.; et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat. Commun. 2015, 6, 8470. [Google Scholar] [CrossRef] [PubMed]
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.; Rowe, M.; Murray, P.G.; et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 2011, 30, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.N.; Zhang, M.Y.; Sheng, M.Y.; Zhang, P.; Chen, Z.Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J. Cancer Res. Clin. 2018, 144, 1065–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallaney, C.; Ostrander, E.L.; Celik, H.; Kramer, A.C.; Martens, A.; Kothari, A.; Koh, W.K.; Haussler, E.; Iwamori, N.; Gontarz, P.; et al. Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 2019, 33, 2506–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lim, K.G.; Feng, M.; Bao, Y.; Lee, P.L.; Cai, Y.; Chen, Y.; Zhang, H.; Marzese, D.; Hoon, D.S.B.; et al. KDM6B Counteracts EZH2-Mediated Suppression of IGFBP5 to Confer Resistance to PI3K/AKT Inhibitor Treatment in Breast Cancer. Mol. Cancer Ther. 2018, 17, 1973–1983. [Google Scholar] [CrossRef] [Green Version]
- Lagunas-Rangel, F.A. KDM6B (JMJD3) and its dual role in cancer. Biochimie 2021, 184, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lwin, T.; Silva, A.; Shah, B.; Tao, J.; Fang, B.; Zhang, L.; Fu, K.; Bi, C.; Li, J.; et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun. 2017, 8, 14920. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Burger, J.A. Erratum to: PI3K Signaling in Normal B Cells and Chronic Lymphocytic Leukemia (CLL). Curr. Top. Microbiol. Immunol. 2016, 393, E1. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Katz, B.Z.; Lipsky, A.; Wiestner, A. Biology of chronic lymphocytic leukemia in different microenvironments: Clinical and therapeutic implications. Hematol. Oncol. Clin. N. Am. 2013, 27, 173–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangle, J.M.; Roberts, T.M.; Zhao, J.J. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.M. The presence of acetyl groups of histones. Biochem. J. 1963, 87, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I.; Poreba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Maldonado, V.; Garcia-Morillo, M.; Delgado, J. The biology behind PI3K inhibition in chronic lymphocytic leukaemia. Ther. Adv. Hematol. 2015, 6, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Kurtova, A.V.; Tamayo, A.T.; Ford, R.J.; Burger, J.A. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): Importance for interactions with the stromal microenvironment and specific targeting. Blood 2009, 113, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Sakamaki, J.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Niimi, K.; Kohara, M.; Sedoh, E.; Fukumoto, M.; Shibata, S.; Sawano, T.; Tashiro, F.; Miyazaki, S.; Kubota, Y.; Miyazaki, J.I.; et al. FOXO1 regulates developmental lymphangiogenesis by upregulating CXCR4 in the mouse-tail dermis. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.R.; Li, X.; Liao, L.P.; Han, J.; Huang, J.; Li, J.C.; Tao, H.R.; Fan, S.J.; Chen, Z.F.; Li, Q.; et al. P300/CBP inhibition sensitizes mantle cell lymphoma to PI3Kdelta inhibitor idelalisib. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, H.; Xu, Z.; Liu, Q.; Liang, Y.; Wang, L.; Cai, X.; Zhang, L.; Hu, L.; Wang, G.; et al. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell Signal 2007, 19, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, M.; Jurkowska, R.Z.; Jurkowski, T.P. Target specificity of mammalian DNA methylation and demethylation machinery. Org. Biomol. Chem. 2018, 16, 1419–1435. [Google Scholar] [CrossRef]
- Zhao, H.F.; Zhang, L.; Guo, S.Q.; Yuan, T.; Xia, B.; Zhang, L.Y.; Zhang, Y.Z. Overexpression of DNA methyltransferase 1 as a negative independent prognostic factor in primary gastrointestinal diffuse large B-cell lymphoma treated with CHOP-like regimen and rituximab. Oncol. Lett. 2015, 9, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Chartomatsidou, E.; Ntoufa, S.; Kotta, K.; Rovida, A.; Akritidou, M.A.; Belloni, D.; Ferrero, E.; Trangas, T.; Stavroyianni, N.; Anagnostopoulos, A.; et al. Inhibition of EZH2 and immune signaling exerts synergistic antitumor effects in chronic lymphocytic leukemia. Blood Adv. 2019, 3, 1891–1896. [Google Scholar] [CrossRef]
- Szurian, K.; Csala, I.; Marosvari, D.; Rajnai, H.; Derso, K.; Bodor, C.; Piurko, V.; Matolcsy, A.; Reiniger, L. EZH2 is upregulated in the proliferation centers of CLL/SLL lymph nodes. Exp. Mol. Pathol. 2018, 105, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Gaiti, F.; Lu, S.X.; Brand, R.M.; Kulm, S.; Chaligne, R.; Gu, H.C.; Huang, K.Y.; Stamenova, E.K.; Beguelin, W.; et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Almaden, J.V.; Liu, Y.C.; Yang, E.; Otero, D.C.; Birnbaum, H.; Davis-Turak, J.; Asagiri, M.; David, M.; Goldrath, A.W.; Hoffmann, A. B-cell survival and development controlled by the coordination of NF-kappaB family members RelB and cRel. Blood 2016, 127, 1276–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, P.; Wang, W.; Wallach, D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity 2004, 21, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neo, W.H.; Lim, J.F.; Grumont, R.; Gerondakis, S.; Su, I.H. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J. Biol. Chem. 2014, 289, 31693–31707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, B.S.; Spurgeon, S.E.; Furman, R.R.; Flinn, I.W.; Coutre, S.E.; Brown, J.R.; Benson, D.M.; Byrd, J.C.; Peterman, S.; Cho, Y.; et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Okkenhaug, K.; Burger, J.A. PI3K Signaling in Normal B Cells and Chronic Lymphocytic Leukemia (CLL). Curr. Top. Microbiol. Immunol. 2016, 393, 123–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rooij, M.F.; Kuil, A.; Kater, A.P.; Kersten, M.J.; Pals, S.T.; Spaargaren, M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: A rationale for combination therapy. Blood 2015, 125, 2306–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondello, P.; Derenzini, E.; Asgari, Z.; Philip, J.; Brea, E.J.; Seshan, V.; Hendrickson, R.C.; de Stanchina, E.; Scheinberg, D.A.; Younes, A. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget 2017, 8, 14017–14028. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Dysregulated Epigenetic Mark in MCL | Gene/Genes Mediating Epigenetic Dysregulation in MCL | Type of Dysregulation | Target Gene/Genes Affected by Epigenetic Dysregulation | Putative Role in MCL | Reference |
|---|---|---|---|---|---|
| DNA methylation | - | DNA hypomethylation at promoter region of target gens | Mediating the expression of SOX11 | Promotes oncogenic cell proliferation | [63] |
| DNA methylation | DNMT1 upregulation | Global DNA hypermethylation | Abnormal expression of β-catenin which upregulates the expression of c-MYC and MMP7 (Matrix Metallopeptidase 7) Reduced expression of tumor suppressor gene; PARG1 | Promotes tumor cell proliferation and survival | [64,65] |
| Histone H3K27acetylation | Mutation in the component of SWI-SNF complex (SMARCA4) | Abnormal histone H3K27 acetylation and chromatin accessibility at the promoter/enhancer region of target genes | Reduced chromatin accessibility at promoter region of transcription factor ATF3 (negative regulator of anti-apoptotic gene BCL-xL) | MCL cell survival and drug resistance | [32,66,67] |
| Global histone acetylation | Abnormal activity of HDACs, Class I, II, e.g., HDAC8 | Enhanced HDAC (Histone deacetylase) activity leading to abnormal histone acetylation and chromatin accessibility | Reduced transcription of pro-apoptotic genes (BIM, BMF) Enhanced expression of c-MYC and PLK1 | Inhibits apoptosis and promotes tumor cell proliferation and survival | [68,69,70,71] |
| Histone H3K36me3 | Gain of function mutation in histone methyltransferase WHSC1 (MMSET) | Enhanced H3K36me3 levels | Enhanced expression of cell cycle regulators | Promotes tumor cell proliferation | [72,73] |
| Histone H3K4 methylation | Loss of function mutation in histone methyltransferase MLL2 (KMT2D) | Diminished H3K4 methylation levels | Functional consequences in MCL are not well understood Contributes to genome instability and transcriptional stress | Disturbs the expression of genes that sustain proliferation and cell survival | [32,74,75,76] |
| H3K27me3 | EZH2 upregulation | Enhanced H3K27me3 levels | Repressed expression of CDKN2B, HOX genes | Promotes MCL cell growth | [77,78] |
| H3K27me3 | KDM6B histone demethylase | Enhanced KDM6B levels, reduces H3K27me3 at promoter region of target genes | Functional consequences in MCL are not well understood In other B-cell malignancies target NF-κB subunits and target genes | Promoters tumor cells survival and drug resistance | [79,80,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeghi, L.; Wright, A.P. Migration and Adhesion of B-Lymphocytes to Specific Microenvironments in Mantle Cell Lymphoma: Interplay between Signaling Pathways and the Epigenetic Landscape. Int. J. Mol. Sci. 2021, 22, 6247. https://doi.org/10.3390/ijms22126247
Sadeghi L, Wright AP. Migration and Adhesion of B-Lymphocytes to Specific Microenvironments in Mantle Cell Lymphoma: Interplay between Signaling Pathways and the Epigenetic Landscape. International Journal of Molecular Sciences. 2021; 22(12):6247. https://doi.org/10.3390/ijms22126247
Chicago/Turabian StyleSadeghi, Laia, and Anthony P. Wright. 2021. "Migration and Adhesion of B-Lymphocytes to Specific Microenvironments in Mantle Cell Lymphoma: Interplay between Signaling Pathways and the Epigenetic Landscape" International Journal of Molecular Sciences 22, no. 12: 6247. https://doi.org/10.3390/ijms22126247
APA StyleSadeghi, L., & Wright, A. P. (2021). Migration and Adhesion of B-Lymphocytes to Specific Microenvironments in Mantle Cell Lymphoma: Interplay between Signaling Pathways and the Epigenetic Landscape. International Journal of Molecular Sciences, 22(12), 6247. https://doi.org/10.3390/ijms22126247