
Behavioral and Neuronal Effects of Inhaled Bromine Gas: Oxidative Brain Stem Damage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
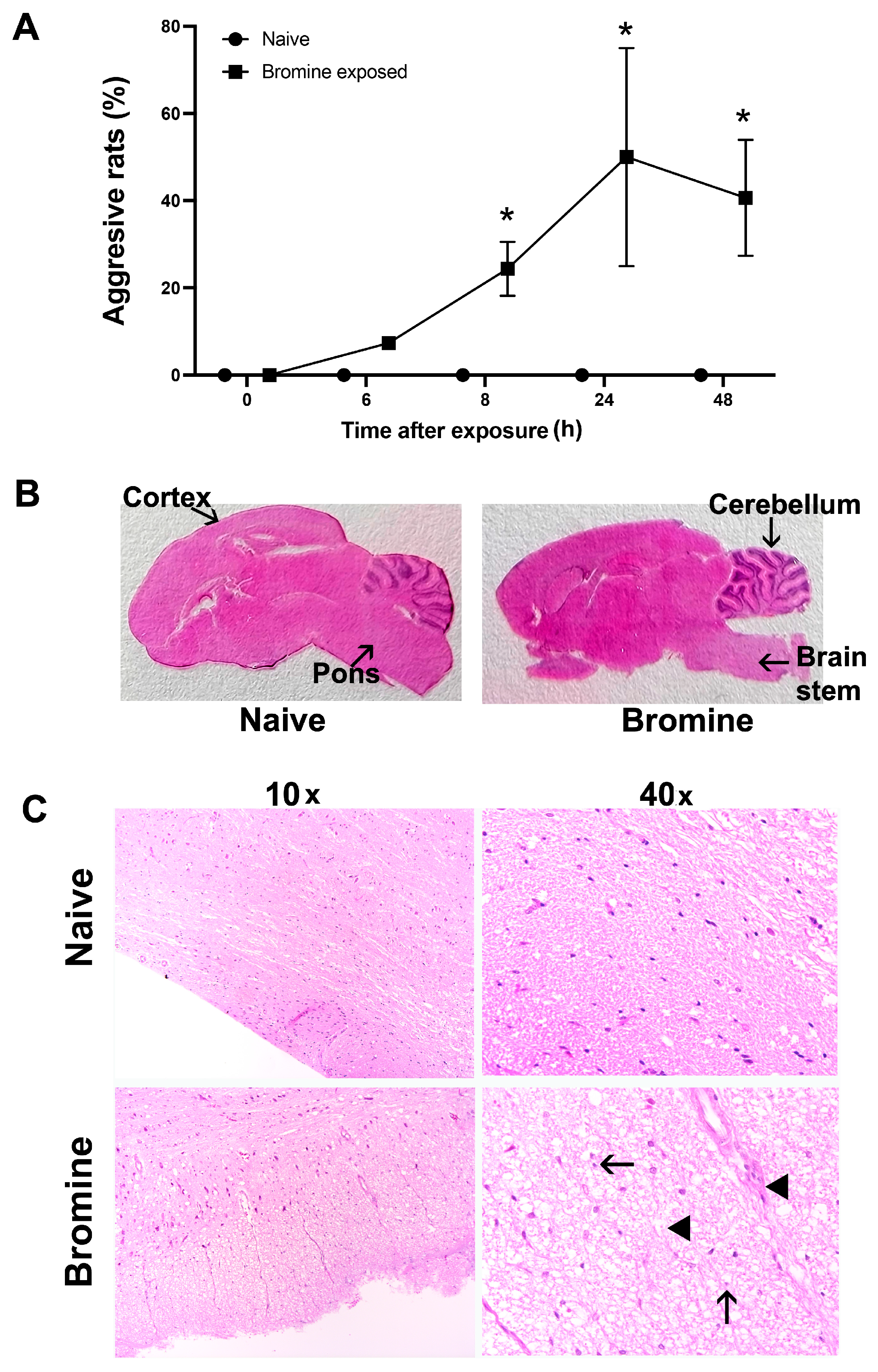
2.1. Altered Cage Behavior Characteristics and Related Brain Injury Induced By Acute Br2 Exposure
2.2. Br2 Increases Brain Injury Biomarker GFAP and Elevates Oxidative Stress in the Brain Stem
2.3. Effect of Br2 Exposure on Aromatic Amino Acid Hydroxylases (AAAHs)
2.4. Br2 Exposure Alters Levels of Brain Stem Catecholamines
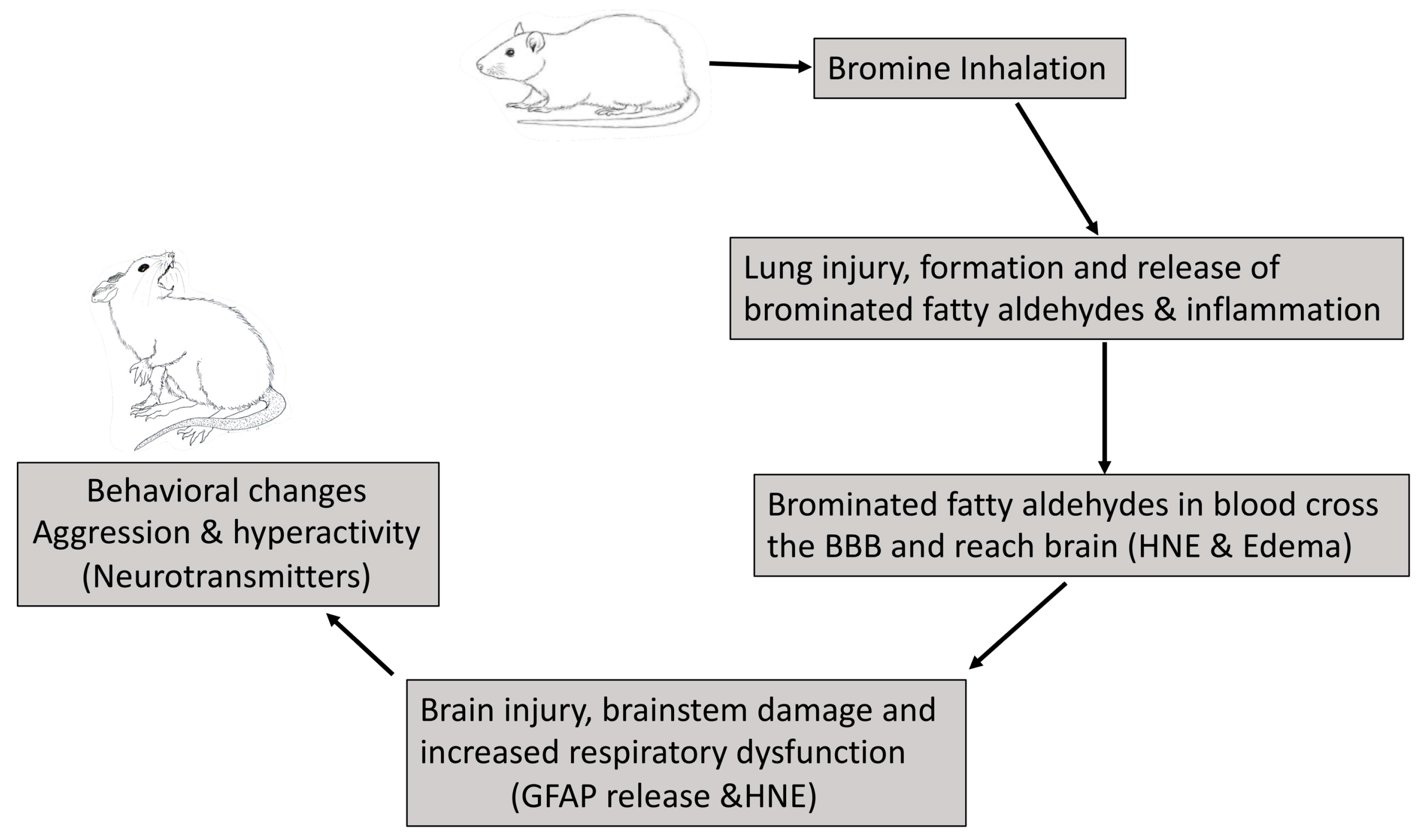
3. Discussion
4. Materials and Methods
4.1. Animals, Bromine Exposure and Cage Behavior Assessment
4.2. Histology and Immunofluorescence
4.3. Western Blots
4.4. Neurochemical Analysis
4.5. Data Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Juncos, J.X.M.; Shakil, S.; Bradley, W.E.; Wei, C.-C.; Zafar, I.; Powell, P.; Mariappan, N.; Louch, W.E.; Ford, D.A.; Ahmad, A.; et al. Chronic cardiac structural damage, diastolic and systolic dysfunction following acute myocardial injury due to bromine exposure in rats. Arch. Toxicol. 2021, 95, 179–193. [Google Scholar] [CrossRef]
- Ahmad, S.; Juncos, J.X.M.; Ahmad, A.; Zaky, A.; Wei, C.-C.; Bradley, W.E.; Zafar, I.; Powell, P.; Mariappan, N.; Vetal, N.; et al. Bromine inhalation mimics ischemia-reperfusion cardiomyocyte injury and calpain activation in rats. Am. J. Physiol. Circ. Physiol. 2019, 316, H212–H223. [Google Scholar] [CrossRef]
- Makarovsky, I.; Markel, G.; Hoffman, A.; Schein, O.; Brosh-Nissimov, T.M.; Finkelstien, A.; Tashma, Z.; Dushnitsky, T.; Eisenkraft, A. Bromine—The red cloud approaching. Isr. Med. Assoc. J. IMAJ 2007, 9, 677–679. [Google Scholar] [PubMed]
- Kothari, P.; Sarda, D.; Gursev; Joshi, P.; Ahmad, A. Cystic nephroma in childhood. Afr. J. Paediatr. Surg. 2009, 6, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Juncos, J.X.M.; Shakil, S.; Ahmad, A.; Aishah, D.; Morgan, C.J.; Dell’Italia, L.J.; Ford, D.A.; Ahmad, A.; Ahmad, S. Circulating and tissue biomarkers as predictors of bromine gas inhalation. Ann. N. Y. Acad. Sci. 2020, 1480, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Gaut, J.P.; Yeh, G.C.; Tran, H.D.; Byun, J.; Henderson, J.P.; Richter, G.M.; Brennan, M.-L.; Lusis, A.J.; Belaaouaj, A.; Hotchkiss, R.S.; et al. Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc. Natl. Acad. Sci. USA 2001, 98, 11961–11966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Public Health England. Bromine Toxicology Overview; Prepared by the Toxicology Department CRCE, PHE; Public Health England: London, UK, 2009. [Google Scholar]
- Haghighi, K.; Bidwell, P.; Kranias, E.G. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 2014, 77, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Kranias, E.G.; Doevendans, P.A.; Glijnis, P.C.; Hajjar, R.J. PLN Foundation. Circ. Res. 2018, 123, 1276–1278. [Google Scholar] [CrossRef]
- Lumpkins, K.; Bochicchio, G.V.; Keledjian, K.; Simard, J.M.; McCunn, M.; Scalea, T. Glial Fibrillary Acidic Protein is Highly Correlated with Brain Injury. J. Trauma Inj. Infect. Crit. Care 2008, 65, 778–784. [Google Scholar] [CrossRef]
- Zhang, D.; Dhillon, H.S.; Mattson, M.P.; Yurek, D.M.; Prasad, R.M. Immunohistochemical detection of the lipid peroxidation product 4-hydroxynonenal after experimental brain injury in the rat. Neurosci. Lett. 1999, 272, 57–61. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free. Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Guo, T.; Ren, P.; Li, X.; Luo, T.; Gong, Y.; Hao, S.; Wang, B. Neural Injuries Induced by Hydrostatic Pressure Associated with Mass Effect after Intracerebral Hemorrhage. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Waløen, K.; Kleppe, R.; Martinez, A.; Haavik, J. Tyrosine and tryptophan hydroxylases as therapeutic targets in human disease. Expert Opin. Ther. Targets 2016, 21, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.; Vetal, N.; Matalon, S.; Aggarwal, S. Role of heme in bromine-induced lung injury. Ann. N. Y. Acad. Sci. 2016, 1374, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, E.; De Kleijn, R.; Colzato, L.S.; Alkemade, A.; Forstmann, B.U.; Nieuwenhuis, S. Neurotransmitters as food supplements: The effects of GABA on brain and behavior. Front. Psychol. 2015, 6, 1520. [Google Scholar] [CrossRef] [Green Version]
- Bembea, M.M.; Savage, W.; Strouse, J.J.; Schwartz, J.M.; Graham, E.; Thompson, C.B.; Everett, A. Glial fibrillary acidic protein as a brain injury biomarker in children undergoing extracorporeal membrane oxygenation*. Pediatr. Crit. Care Med. 2011, 12, 572–579. [Google Scholar] [CrossRef]
- Marcelis, M.; Suckling, J.; Hofman, P.; Woodruff, P.; Bullmore, E.; Van Os, J. Evidence that brain tissue volumes are associated with HVA reactivity to metabolic stress in schizophrenia. Schizophr. Res. 2006, 86, 45–53. [Google Scholar] [CrossRef]
- Salvi, A.; Salim, S. Neurobehavioral Consequences of Traffic-Related Air Pollution. Front. Neurosci. 2019, 13, 1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, R.D. Handbook of Drug and Chemical Stimulation of the Brain: Behavioral, Pharmacological, and Physiological Aspects; Van Nostrand Reinhold Co.: New York, NY, USA, 1974. [Google Scholar]
- Zhong, Z.; Zeng, T.; Xie, K.; Zhang, C.; Chen, J.; Bi, Y.; Zhao, X. Elevation of 4-hydroxynonenal and malondialdehyde modified protein levels in cerebral cortex with cognitive dysfunction in rats exposed to 1-bromopropane. Toxicology 2013, 306, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Patki, G.; Atrooz, F.; Alkadhi, I.; Solanki, N.; Salim, S. High aggression in rats is associated with elevated stress, anxiety-like behavior, and altered catecholamine content in the brain. Neurosci. Lett. 2015, 584, 308–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patki, G.; Solanki, N.; Salim, S. Witnessing traumatic events causes severe behavioral impairments in rats. Int. J. Neuropsychopharmacol. 2014, 17, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Patki, G.; Solanki, N.; Atrooz, F.; Ansari, A.; Allam, F.; Jannise, B.; Maturi, J.; Salim, S. Novel mechanistic insights into treadmill exercise based rescue of social defeat-induced anxiety-like behavior and memory impairment in rats. Physiol. Behav. 2014, 130, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Lam, A.; Bolisetty, S.; Carlisle, M.A.; Traylor, A.; Agarwal, A.; Matalon, S. Heme Attenuation Ameliorates Irritant Gas Inhalation-Induced Acute Lung Injury. Antioxid. Redox Signal. 2016, 24, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Miczek, K.A. Neurogenetics of Aggressive Behavior: Studies in Rodents. Curr. Top. Behav. Neurosci. 2013, 17, 3–44. [Google Scholar] [CrossRef] [Green Version]
- Vos, P.E.; Jacobs, B.; Andriessen, T.M.J.C.; Lamers, K.J.B.; Borm, G.F.; Beems, T.; Edwards, M.; Rosmalen, C.F.; Vissers, J.L.M. GFAP and S100B are biomarkers of traumatic brain injury: An observational cohort study. Neurology 2010, 75, 1786–1793. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Daughtrey, W.C.; Clark, C.R.; Schreiner, C.A.; White, R. Health assessment of gasoline and fuel oxygenate vapors: Neurotoxicity evaluation. Regul. Toxicol. Pharmacol. 2014, 70, S35–S42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantharam, P.; Whitley, E.M.; Mahama, B.; Kim, D.S.; Imerman, P.M.; Shao, D.; Langley, M.R.; Kanthasamy, A.; Rumbeiha, W.K. Characterizing a mouse model for evaluation of countermeasures against hydrogen sulfide-induced neuro-toxicity and neurological sequelae. Ann. N. Y. Acad. Sci. 2017, 1400, 46–54. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Kinetics of Hypobromous Acid-Mediated Oxidation of Lipid Components and Antioxidants. Chem. Res. Toxicol. 2007, 20, 1980–1988. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Kinetic Analysis of the Reactions of Hypobromous Acid with Protein Components: Implications for Cellular Damage and Use of 3-Bromotyrosine as a Marker of Oxidative Stress†. Biochemistry 2004, 43, 4799–4809. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, E.J.; McGill, C.M.; Barth, B.M. Sphingolipids as Regulators of Neuro-Inflammation and NADPH Oxidase 2. Neuro Mol. Med. 2021, 23, 25–46. [Google Scholar] [CrossRef]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.; Lee, S.; Lim, S.; Noh, J.; Cho, S.Y.; Ha, J.; Kim, H.; Kim, C.; Park, S.; et al. Exposure of ultrafine particulate matter causes glutathione redox imbalance in the hippocampus: A neurometabolic susceptibility to Alzheimer’s pathology. Sci. Total Environ. 2020, 718, 137267. [Google Scholar] [CrossRef] [PubMed]
- Nolan, R.; Gaskill, P. The role of catecholamines in HIV neuropathogenesis. Brain Res. 2019, 1702, 54–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Huang, E.Y.; Kuo, T.T.; Miller, J.; Chiang, Y.H.; Hoffer, B.J. Impact of Traumatic Brain Injury on Dopaminergic Transmission. Cell Transpl. 2017, 26, 1156–1168. [Google Scholar] [CrossRef] [Green Version]
- Sofic, E.; Denisova, N.; Youdim, K.; Vatrenjak-Velagic, V.; De Filippo, C.; Mehmedagic, A.; Causevic, A.; Cao, G.; Joseph, J.A.; Prior, R.L. Antioxidant and pro-oxidant capacity of catecholamines and related compounds. Effects of hydrogen peroxide on glutathione and sphingomyelinase activity in pheochromocytoma PC12 cells: Potential relevance to age-related diseases. J. Neural Transm. 2001, 108, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, L.M.; Sullivan, P.; Dunn, A.R.; Bermejo, M.K.; Fu, R.; Masoud, S.T.; Gregersen, E.; Urs, N.M.; Nazari, R.; Jensen, P.H.; et al. Enhanced tyrosine hydroxylase activity induces oxidative stress, causes accumulation of autotoxic catecholamine metabolites, and augments amphetamine effects in vivo. J. Neurochem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Castrén, E.; Kasper, S.; Lanzenberger, R. Serotonin and neuroplasticity—Links between molecular, functional and structural pathophysiology in depression. Neurosci. Biobehav. Rev. 2017, 77, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffei, M.E. 5-Hydroxytryptophan (5-HTP): Natural Occurrence, Analysis, Biosynthesis, Biotechnology, Physiology and Toxicology. Int. J. Mol. Sci. 2020, 22, 181. [Google Scholar] [CrossRef]
- Wu, H.; Denna, T.H.; Storkersen, J.N.; Gerriets, V.A. Beyond a neurotransmitter: The role of serotonin in inflammation and immunity. Pharmacol. Res. 2019, 140, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.S.; Muresanu, D.F.; Nozari, A.; Castellani, R.J.; Dey, P.K.; Wiklund, L.; Sharma, A. Chapter Two—Anesthetics influence concussive head injury induced blood-brain barrier breakdown, brain edema formation, cerebral blood flow, serotonin levels, brain pathology and functional outcome. Int. Rev. Neurobiol. 2019, 146, 45–81. [Google Scholar] [CrossRef] [PubMed]
- Belkacemi, L.; Darmani, N.A. Dopamine receptors in emesis: Molecular mechanisms and potential therapeutic function. Pharmacol. Res. 2020, 161, 105124. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Xing, L.; Yang, X.; Xu, J.; Ren, Q.; Su, K.-P.; Lu, Y.; Wang, F. Association of Cigarette Smoking with Sleep Disturbance and Neurotransmitters in Cerebrospinal Fluid. Nat. Sci. Sleep 2020, ume 12, 801–808. [Google Scholar] [CrossRef]
- Mackie, E.; Svendsen, E.; Grant, S.; Michels, J.E.; Richardson, W.H. Management of Chlorine Gas-Related Injuries from the Graniteville, South Carolina, Train Derailment. Disaster Med. Public Heal. Prep. 2014, 8, 411–416. [Google Scholar] [CrossRef]
- Ghosh, A.; Langley, M.R.; Harischandra, D.S.; Neal, M.L.; Jin, H.; Anantharam, V.; Joseph, J.; Brenza, T.; Narasimhan, B.; Kanthasamy, A.; et al. Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Pre-clinical Animal Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2016, 11, 259–278. [Google Scholar] [CrossRef] [Green Version]
- Anantharam, P.; Whitley, E.M.; Mahama, B.; Kim, D.; Sarkar, S.; Santana, C.; Chan, A.; Kanthasamy, A.; Kanthasamy, A.; Boss, G.R.; et al. Cobinamide is effective for treatment of hydrogen sulfide-induced neurological sequelae in a mouse model. Ann. N. Y. Acad. Sci. 2017, 1408, 61–78. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakil, S.; Masjoan Juncos, J.X.; Mariappan, N.; Zafar, I.; Amudhan, A.; Amudhan, A.; Aishah, D.; Siddiqui, S.; Manzoor, S.; Santana, C.M.; et al. Behavioral and Neuronal Effects of Inhaled Bromine Gas: Oxidative Brain Stem Damage. Int. J. Mol. Sci. 2021, 22, 6316. https://doi.org/10.3390/ijms22126316
Shakil S, Masjoan Juncos JX, Mariappan N, Zafar I, Amudhan A, Amudhan A, Aishah D, Siddiqui S, Manzoor S, Santana CM, et al. Behavioral and Neuronal Effects of Inhaled Bromine Gas: Oxidative Brain Stem Damage. International Journal of Molecular Sciences. 2021; 22(12):6316. https://doi.org/10.3390/ijms22126316
Chicago/Turabian StyleShakil, Shazia, Juan Xavier Masjoan Juncos, Nithya Mariappan, Iram Zafar, Apoorva Amudhan, Archita Amudhan, Duha Aishah, Simmone Siddiqui, Shajer Manzoor, Cristina M. Santana, and et al. 2021. "Behavioral and Neuronal Effects of Inhaled Bromine Gas: Oxidative Brain Stem Damage" International Journal of Molecular Sciences 22, no. 12: 6316. https://doi.org/10.3390/ijms22126316
APA StyleShakil, S., Masjoan Juncos, J. X., Mariappan, N., Zafar, I., Amudhan, A., Amudhan, A., Aishah, D., Siddiqui, S., Manzoor, S., Santana, C. M., Rumbeiha, W. K., Salim, S., Ahmad, A., & Ahmad, S. (2021). Behavioral and Neuronal Effects of Inhaled Bromine Gas: Oxidative Brain Stem Damage. International Journal of Molecular Sciences, 22(12), 6316. https://doi.org/10.3390/ijms22126316