
Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy

 , ,
, ,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
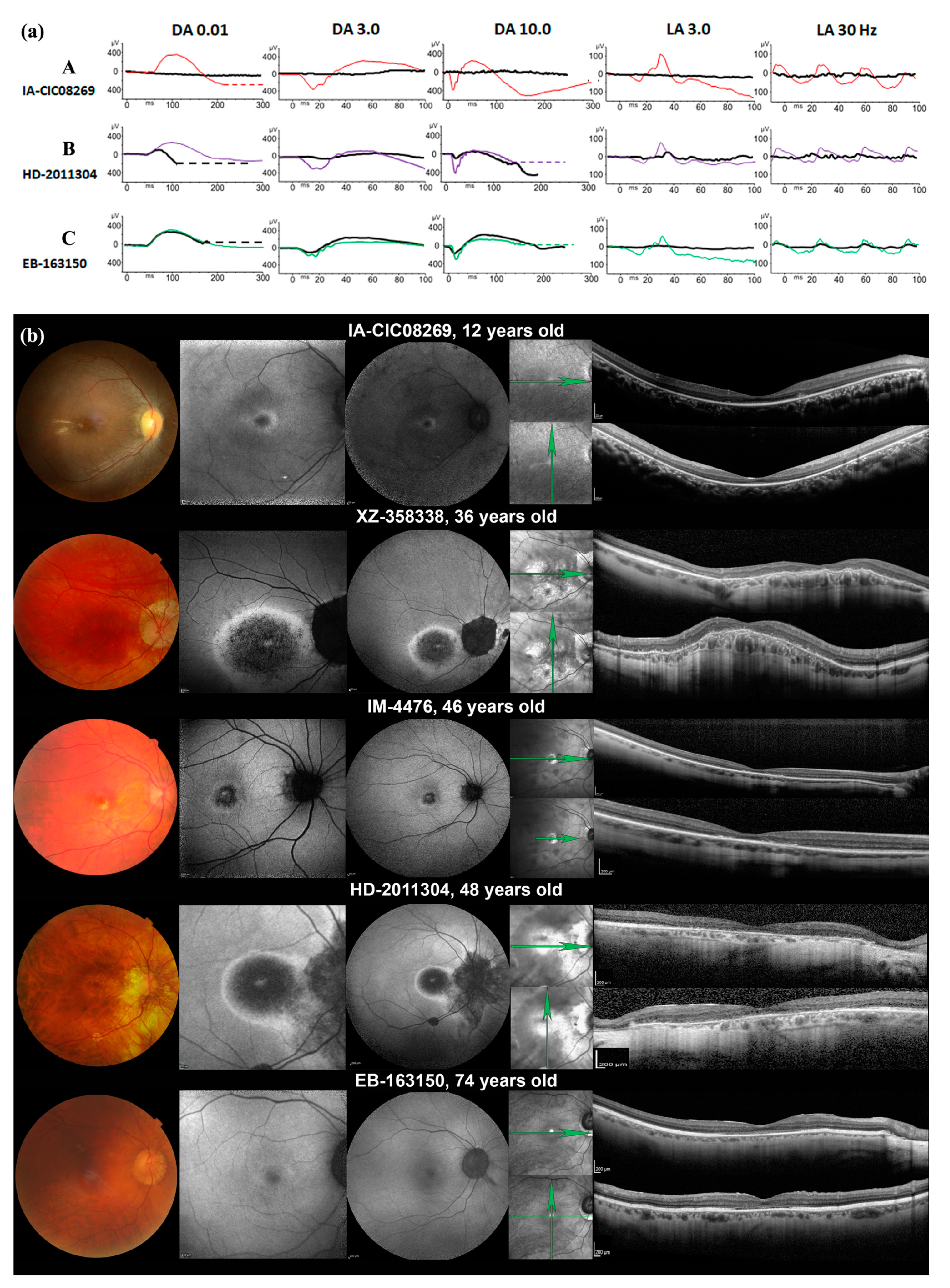
2.1. Phenotypic Characterization
2.2. Genotyping Results
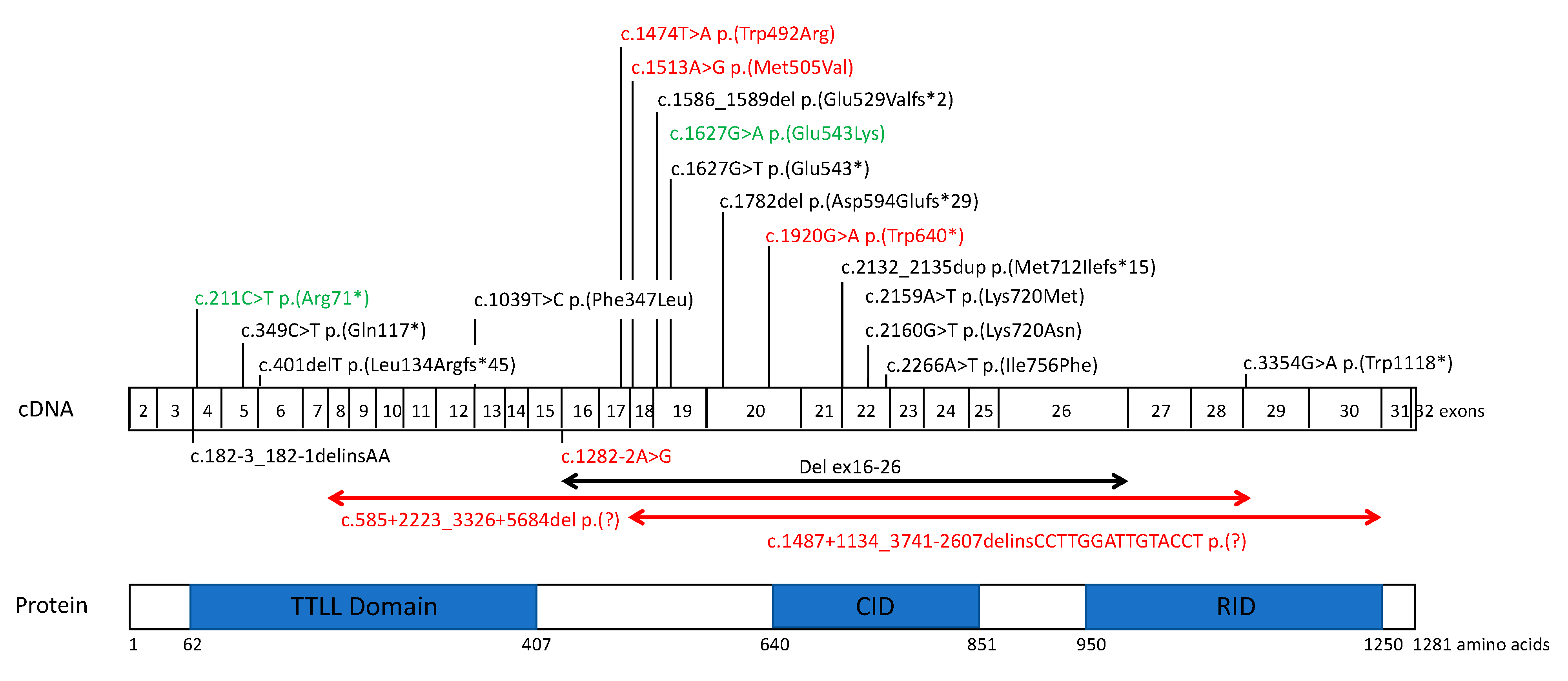
2.3. CNV Identification and Breakpoint Characterization
3. Discussion
3.1. Phenotype Characteristics
3.2. TTLL5 Variants and Structure-Function Correlation
3.3. Genotype-Phenotype Correlations
3.4. Future Directions
4. Materials and Methods
4.1. Subject and Clinical Examination
4.2. Genetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamel, C.P. Cone Rod Dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive Cone and Cone-Rod Dystrophies: Phenotypes and Underlying Molecular Genetic Basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.H.J.; Phan, T.M.L.; Zekveld-Vroon, R.C.; Leroy, B.P.; van den Born, L.I.; Hoyng, C.B.; Klaver, C.C.W.; Writing Committee for the Cone Disorders Study Group Consortium; Roosing, S.; Pott, J.-W.R.; et al. Clinical Course, Genetic Etiology, and Visual Outcome in Cone and Cone-Rod Dystrophy. Ophthalmology 2012, 119, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The Cone Dysfunction Syndromes. Br. J. Ophthalmol. 2004, 88, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Boulanger-Scemama, E.; El Shamieh, S.; Démontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.-P.; Letexier, M.; Souied, E.; et al. Next-Generation Sequencing Applied to a Large French Cone and Cone-Rod Dystrophy Cohort: Mutation Spectrum and New Genotype-Phenotype Correlation. Orphanet J. Rare Dis. 2015, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive Cone and Cone-Rod Dystrophies: Clinical Features, Molecular Genetics and Prospects for Therapy. Br. J. Ophthalmol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Sharma, T. Progressive Cone Dystrophy and Cone-Rod Dystrophy (XL, AD, and AR). Adv. Exp. Med. Biol. 2018, 1085, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sergouniotis, P.I.; Chakarova, C.; Murphy, C.; Becker, M.; Lenassi, E.; Arno, G.; Lek, M.; MacArthur, D.G.; UCL-Exomes Consortium; Bhattacharya, S.S.; et al. Biallelic Variants in TTLL5, Encoding a Tubulin Glutamylase, Cause Retinal Dystrophy. Am. J. Hum. Genet. 2014, 94, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.-S.; He, Y.; Dougherty, E.J.; Jimenez-Movilla, M.; Avella, M.; Grullon, S.; Sharlin, D.S.; Guo, C.; Blackford, J.A.; Awasthi, S.; et al. Disruption of Ttll5/Stamp Gene (Tubulin Tyrosine Ligase-like Protein 5/SRC-1 and TIF2-Associated Modulatory Protein Gene) in Male Mice Causes Sperm Malformation and Infertility. J. Biol. Chem. 2013, 288, 15167–15180. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, J.; Miro, J.; Strub, J.-M.; Lacroix, B.; van Dorsselaer, A.; Edde, B.; Janke, C. Polyglutamylation Is a Post-Translational Modification with a Broad Range of Substrates. J. Biol. Chem. 2008, 283, 3915–3922. [Google Scholar] [CrossRef] [Green Version]
- Mahalingan, K.K.; Keith Keenan, E.; Strickland, M.; Li, Y.; Liu, Y.; Ball, H.L.; Tanner, M.E.; Tjandra, N.; Roll-Mecak, A. Structural Basis for Polyglutamate Chain Initiation and Elongation by TTLL Family Enzymes. Nat. Struct. Mol. Biol. 2020, 27, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Park, J.H.; Gumerson, J.; Wu, Z.; Swaroop, A.; Qian, H.; Roll-Mecak, A.; Li, T. Loss of RPGR Glutamylation Underlies the Pathogenic Mechanism of Retinal Dystrophy Caused by TTLL5 Mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E2925–E2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoni, N.; Haer-Wigman, L.; Vaclavik, V.; Tran, V.H.; Farinelli, P.; Balzano, S.; Royer-Bertrand, B.; El-Asrag, M.E.; Bonny, O.; Ikonomidis, C.; et al. Mutations in the Polyglutamylase Gene TTLL5, Expressed in Photoreceptor Cells and Spermatozoa, Are Associated with Cone-Rod Degeneration and Reduced Male Fertility. Hum. Mol. Genet. 2016, ddw282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.d.S.; Hamel, C.P.; Meunier, I.; Varin, J.; Blanchard, S.; Boyard, F.; Sahel, J.-A.; Zeitz, C. Novel Splice-Site Mutation in TTLL5 Causes Cone Dystrophy in a Consanguineous Family. Mol. Vis. 2017, 23, 131–139. [Google Scholar] [PubMed]
- Méjécase, C.; Kozak, I.; Moosajee, M. The Genetic Landscape of Inherited Eye Disorders in 74 Consecutive Families from the United Arab Emirates. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 762–772. [Google Scholar] [CrossRef]
- Patel, N.; Alkuraya, H.; Alzahrani, S.S.; Nowailaty, S.R.; Seidahmed, M.Z.; Alhemidan, A.; Ben-Omran, T.; Ghazi, N.G.; Al-Aqeel, A.; Al-Owain, M.; et al. Mutations in Known Disease Genes Account for the Majority of Autosomal Recessive Retinal Dystrophies. Clin. Genet. 2018, 94, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-Number Variation Contributes 9% of Pathogenicity in the Inherited Retinal Degenerations. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive Genetic Testing in the Clinical Evaluation of 1119 Patients with Hearing Loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [Green Version]
- van Schooneveld, M.J.; Went, L.N.; Oosterhuis, J.A. Dominant Cone Dystrophy Starting with Blue Cone Involvement. Br. J. Ophthalmol. 1991, 75, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Kohl, S.; Llavona, P.; Sauer, A.; Reuter, P.; Weisschuh, N.; Kempf, M.; Dehmelt, F.A.; Arrenberg, A.B.; Sliesoraityte, I.; Zrenner, E.; et al. A Duplication on Chromosome 16q12 Affecting the IRXB Gene Cluster Is Associated with Autosomal Dominant Cone Dystrophy with Early Tritanopic Color Vision Defect. Hum. Mol. Genet. 2021. [Google Scholar] [CrossRef]
- Pinckers, A. Dominant Cone Dystrophy Starting with Blue Cone Involvement. Br. J. Ophthalmol. 1992, 76, 127. [Google Scholar] [CrossRef] [Green Version]
- Zrenner, E.; Nowicki, J.; Adamczyk, R. Cone Function and Cone Interaction in Hereditary Degenerations of the Central Retina. Doc. Ophthalmol. 1986, 62, 5–12. [Google Scholar] [CrossRef]
- Simunovic, M.P. Acquired Color Vision Deficiency. Surv. Ophthalmol. 2016, 61, 132–155. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber Congenital Amaurosis/Early-Onset Severe Retinal Dystrophy: Clinical Features, Molecular Genetics and Therapeutic Interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Foxman, S.G.; Heckenlively, J.R.; Bateman, J.B.; Wirtschafter, J.D. Classification of Congenital and Early Onset Retinitis Pigmentosa. Arch. Ophthalmol. Chic. Ill. 1960 1985, 103, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, B.; Gyürüs, P.; Preising, M.; Bremser, D.; Gu, S.; Andrassi, M.; Gerth, C.; Gal, A. Early-Onset Severe Rod-Cone Dystrophy in Young Children with RPE65 Mutations. Invest. Ophthalmol. Vis. Sci. 2000, 41, 2735–2742. [Google Scholar] [PubMed]
- Weleber, R.G.; Michaelides, M.; Trzupek, K.M.; Stover, N.B.; Stone, E.M. The Phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from Mutation of RPE65 and Differentiation from Leber Congenital Amaurosis. Invest. Ophthalmol. Vis. Sci. 2011, 52, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, N.; Pennesi, M.E.; Yang, P.; Trzupek, K.M.; Schlechter, C.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- McGee, T.L.; Seyedahmadi, B.J.; Sweeney, M.O.; Dryja, T.P.; Berson, E.L. Novel Mutations in the Long Isoform of the USH2A Gene in Patients with Usher Syndrome Type II or Non-Syndromic Retinitis Pigmentosa. J. Med. Genet. 2010, 47, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Seyedahmadi, B.J.; Rivolta, C.; Keene, J.A.; Berson, E.L.; Dryja, T.P. Comprehensive Screening of the USH2A Gene in Usher Syndrome Type II and Non-Syndromic Recessive Retinitis Pigmentosa. Exp. Eye Res. 2004, 79, 167–173. [Google Scholar] [CrossRef]
- Méjécase, C.; Hummel, A.; Mohand-Saïd, S.; Andrieu, C.; El Shamieh, S.; Antonio, A.; Condroyer, C.; Boyard, F.; Foussard, M.; Blanchard, S.; et al. Whole Exome Sequencing Resolves Complex Phenotype and Identifies CC2D2A Mutations Underlying Non-Syndromic Rod-Cone Dystrophy. Clin. Genet. 2019, 95, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-J.; Li, J.-K.; Chen, H.; Hu, F.-Y.; Zhang, S.-H.; Qi, Y.-H.; Xu, P.; Wang, D.-D.; Wang, L.-S.; Chang, Q.; et al. Genetic and Clinical Findings in a Large Cohort of Chinese Patients with Suspected Retinitis Pigmentosa. Ophthalmology 2019, 126, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A Nationwide Genetic Analysis of Inherited Retinal Diseases in Israel as Assessed by the Israeli Inherited Retinal Disease Consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, V.; Karali, M.; Melillo, P.; Testa, F.; Brunetti-Pierri, R.; Musacchia, F.; Condroyer, C.; Neidhardt, J.; Audo, I.; Zeitz, C.; et al. Spectrum of Disease Severity in Patients With X-Linked Retinitis Pigmentosa Due to RPGR Mutations. Invest. Ophthalmol. Vis. Sci. 2020, 61, 36. [Google Scholar] [CrossRef] [PubMed]
- Hadalin, V.; Šuštar, M.; Volk, M.; Maver, A.; Sajovic, J.; Jarc-Vidmar, M.; Peterlin, B.; Hawlina, M.; Fakin, A. Cone Dystrophy Associated with a Novel Variant in the Terminal Codon of the RPGR-ORF15. Genes 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-T.-A.; Talib, M.; van Schooneveld, M.J.; Brinks, J.; Ten Brink, J.; Florijn, R.J.; Wijnholds, J.; Verdijk, R.M.; Bergen, A.A.; Boon, C.J.F. RPGR-Associated Dystrophies: Clinical, Genetic, and Histopathological Features. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, L.E.L.M.; Bhatt, S.S.; Janssen, I.M.; Xia, Z.; Lalani, S.R.; Pfundt, R.; Derwinska, K.; de Vries, B.B.A.; Gilissen, C.; Hoischen, A.; et al. Rare Pathogenic Microdeletions and Tandem Duplications Are Microhomology-Mediated and Stimulated by Local Genomic Architecture. Hum. Mol. Genet. 2009, 18, 3579–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schil, K.; Naessens, S.; Van de Sompele, S.; Carron, M.; Aslanidis, A.; Van Cauwenbergh, C.; Mayer, A.K.; Van Heetvelde, M.; Bauwens, M.; Verdin, H.; et al. Correction: Mapping the Genomic Landscape of Inherited Retinal Disease Genes Prioritizes Genes Prone to Coding and Noncoding Copy-Number Variations. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1998. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, J.; Rogowski, K.; Miro, J.; Lacroix, B.; Eddé, B.; Janke, C. A Targeted Multienzyme Mechanism for Selective Microtubule Polyglutamylation. Mol. Cell 2007, 26, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Astuti, G.D.N.; Arno, G.; Hull, S.; Pierrache, L.; Venselaar, H.; Carss, K.; Raymond, F.L.; Collin, R.W.J.; Faradz, S.M.H.; van den Born, L.I.; et al. Mutations in AGBL5, Encoding α-Tubulin Deglutamylase, Are Associated With Autosomal Recessive Retinitis Pigmentosa. Invest. Ophthalmol. Vis. Sci. 2016, 57, 6180–6187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audo, I.; Bujakowska, K.M.; Léveillard, T.; Mohand-Saïd, S.; Lancelot, M.-E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.-P.; Letexier, M.; et al. Development and Application of a Next-Generation-Sequencing (NGS) Approach to Detect Known and Novel Gene Defects Underlying Retinal Diseases. Orphanet J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoffroy, V.; Stoetzel, C.; Scheidecker, S.; Schaefer, E.; Perrault, I.; Bär, S.; Kröll, A.; Delbarre, M.; Antin, M.; Leuvrey, A.-S.; et al. Whole-Genome Sequencing in Patients with Ciliopathies Uncovers a Novel Recurrent Tandem Duplication in IFT140. Hum. Mutat. 2018, 39, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Patient, Sex | Symptoms | BCVA at First Visit | Refraction | Anterior Segment | Fundus | Colour Vision | Visual Field | ffERG | SWAF | SD-OCT |
|---|---|---|---|---|---|---|---|---|---|---|
| IA-CIC08269, F | Night vision difficulties since 6 y.o. Bilateral decreased visual acuity | RE: 20/50 LE: 20/50 7 y.o. | +1.0(−1.5)10° +1.0(−1.5)175° | Normal | Normal optic discs Mild vascular narrowing Dark fovea, yellowish deposit with indistinct borders surrounding the fovea Whitish spots of chorio-retinal atrophy and coarse pigmentary migration in peripheral retina | Tritan defect | Kinetic perimetry: V4e: 145° horizontal and 120° vertical III1e: constricted to 5° I1e: not perceived OU | Undetectable scotopic- and photopic responses | Very narrow ring of increased autofluorescence | Outer retinal layers (EZ, ONL) disappearance outside the fovea |
| XZ-358338, M | Progressive visual loss since teens, Photophobia from age 36 y.o. | RE: 20/50 LE: 20/40 36 y.o. | −12.5(−2.0)0° −12.0(−3.0)160° | Phakic IOL implanted at 36 y.o. | Normal discs Peripapillary atrophy (conus myopicus) Mild arteriolar narrowing Dark round atrophic lesion of the macula with indistinct borders Normal peripheral retina | Tritan defect | Static perimetry: Absolute central scotoma of 5° surrounded by a ring of reduced sensitivity OU | Generalized cone-rod dysfunction (scotopic responses 70% of normal amplitude and photopic responses 25% of normal amplitude) | Salt and pepper round macular lesion with hyperautofluorescent edges Normal autofluorescence in the peripheral retina | Dome-shaped macula Outer retinal layers (EZ, ONL) disappearance in the macula; normal aspect outside the macular lesion |
| IM-4476, M | Progressive visual acuity loss since childhood, Photophobia and rapid visual acuity loss since 42 y.o. | RE:20/100 LE: 20/50 46 y.o. | −6.25(−3.0)170° −6.0(−3.50)35° | Normal | Normal discs Chorioretinal thinning between the macula and optic disc Yellowish atrophic round macular lesion Normal peripheral retina | Severe dyschromatopsia | V4e: 150° horizontal and 120° vertical II4e: 90° horizontal and 60° vertical I4e not perceived OU | Severe generalized cone system dysfunction with normal rod system function | Round foveal hypoautofluorescent lesion with irregular hyperautofluorescent edges | Foveal disappearance of outer retinal layers (EZ, ONL) |
| HD-2011304, M | Night vision disturbances since his forties Photophobia and rapid visual acuity loss since 45 y.o. | RE:20/63 LE:20/50 48 y.o. | -6.75(−3.50)80° −9.50(−3.50)110° | Normal | Normal disc Peripapillary atrophy (conus myopicus) Mild arteriolar narrowing Dark foveal spot surrounded by ovoid zone of macular discoloration Normal peripheral retina | Tritan defect | V4e: 100° horizontal and 90° vertical V4e central scotoma of 15° | Generalized cone-rod dysfunction (scotopic responses 50% of normal amplitude, photopic responses 30% of normal amplitude) | Foveal hyperautofluorescent spot surrounded by a round hypoautofluorescent area with hyperautofluorescent edges Macular lesion is continuous with the peripapillary atrophy | Perifoveal disappearance of outer retinal layers (EZ, ONL) Foveal sparing of EZ |
| EB-163150, M | Progressive visual acuity loss from teens Photophobia since childhood, became disabling from thirties | RE: 20/80 LE: 20/80 70 y.o. | −6.0(−1.0)40° −7.0(−0.75)165° | Cortico-nuclear and posterior subcapsular cataract OU | Normal disc Peripapillary atrophy (conus myopicus) Mild arteriolar narrowing Round macular discoloration | Tritan defect | V4e: 140° horizontal and vertical V1e: central scotoma of 15° Humphrey 10-2: sparing of fixation point, absolute annular scotoma at 5° of eccentricity surrounded by a relative deficit | Severe cone system dysfunction with only residual photopic responses while scotopic responses were normal | Perifoveal hyperautofluorescent ring with indistinct edges | Perifoveal disappearance of outer retinal layers (EZ, ONL) Foveal sparing of EZ |
| Patient ID | Status | Genomic Position | DNA Variant | Protein Variant | Variant Type | GnomAD AF (NFE) | In Silico Prediction | ACMG Rules | ACMG Classification | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IA-CIC08269 | Homozygous | 76232616 | c.1920G>A | p.(Trp640*) | Nonsense | 0 | CADD | 36.0 | PVS1 Very strong | Pathogenic | This study |
| PM2 Moderate | |||||||||||
| PP3 Supporting | |||||||||||
| PP4 Supporting | |||||||||||
| PP5 Supporting | |||||||||||
| XZ-358338 | Compound Heterozygous | 76147917 | c.211C>T | p.(Arg71*) | Nonsense | 0.00002 | CADD | 35.0 | PVS1 Very strong | Pathogenic | Zampaglione 2020 |
| PM2 Moderate | |||||||||||
| PM3 Moderate | |||||||||||
| PP3 Supporting | |||||||||||
| PP4 Supporting | |||||||||||
| PP5 Supporting | |||||||||||
| 76167836_ 76292188 | c.585+2223_3326+5684del | p.(Pro196Glufs*47) | Exons 8-28 deletion | 0 | PVS1 Very strong | Pathogenic | This study | ||||
| PM2 Moderate | |||||||||||
| PM3 Moderate | |||||||||||
| PP3 Supporting | |||||||||||
| PP4 Supporting | |||||||||||
| PP5 Supporting | |||||||||||
| IM-4476 | Compound Heterozygous | 76213058_ 76365878 | c.1487+1134_3741- 2607delins15 | p.(Ser497_Lys1247del) | Exons 18-30 deletion | 0 | PVS1 Very strong | Pathogenic | This study | ||
| PM2 Moderate | |||||||||||
| PP3 Supporting | |||||||||||
| PP4 Supporting | |||||||||||
| PP5 Supporting | |||||||||||
| 76231034 | c.1627G>A | p.(Glu543Lys) | Missense | 0.0003 | Conservation (Grantham) | 56 (0–215) | PS3 Strong | Pathogenic | Sergouniotis 2014 | ||
| CADD | 27.6 | PM2 Moderate | |||||||||
| SIFT | Deleterious (score: 0.02) | PP3 Supporting | |||||||||
| Polyphen2 | Probably Damaging (1.000) | PP4 Supporting | |||||||||
| PP5 Supporting | |||||||||||
| HD-2011304 | Compound Heterozygous | 76211911 | c.1474T>A | p.(Trp492Arg) | Missense | 0 | Conservation (Grantham) | 101 (0–215) | PM2 Moderate | Likely Pathogenic * | This study |
| CADD | 28.3 | PM3 Moderate | |||||||||
| SIFT | Deleterious (score: 0) | PP3 Supporting | |||||||||
| Polyphen2 | Probably Damaging (0.995) | PP4 Supporting | |||||||||
| 76231034 | c.1627G>A | p.(Glu543Lys) | Missense | 0.0003 | Conservation (Grantham) | 56 (0–215) | PS3 Strong | Pathogenic | Sergouniotis 2014 | ||
| CADD | 27.6 | PM2 Moderate | |||||||||
| SIFT | Deleterious (score: 0.02) | PP3 Supporting | |||||||||
| Polyphen2 | Probably Damaging (1.000) | PP4 Supporting | |||||||||
| PP5 Supporting | |||||||||||
| EB-163150 | Compound Heterozygous | 76211436 | c.1282-2A>G | p.(?) | Splicing | 0.000009 | CADD | 34.0 | PVS1 Very strong | Pathogenic | This study |
| SpliceSiteFinder | −100% | PM2 Moderate | |||||||||
| MaxEnt | −100% | PP3 Supporting | |||||||||
| SpliceAI | 0.85 | PP4 Supporting | |||||||||
| 76219261 | c.1513A>G | p.(Met505Val) | Missense | 0.00005 | Conservation (Grantham) | 21 (0–215) | PM2 Moderate | Likely Pathogenic * | This study | ||
| CADD | 23.0 | PM3 Moderate | |||||||||
| SIFT | Tolerated (score: 0.2) | PP3 Supporting | |||||||||
| Polyphen2 | Possibly Damaging (0.786) | PP4 Supporting | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnov, V.; Grunewald, O.; Muller, J.; Zeitz, C.; Obermaier, C.D.; Devos, A.; Pelletier, V.; Bocquet, B.; Andrieu, C.; Bacquet, J.-L.; et al. Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy. Int. J. Mol. Sci. 2021, 22, 6410. https://doi.org/10.3390/ijms22126410
Smirnov V, Grunewald O, Muller J, Zeitz C, Obermaier CD, Devos A, Pelletier V, Bocquet B, Andrieu C, Bacquet J-L, et al. Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy. International Journal of Molecular Sciences. 2021; 22(12):6410. https://doi.org/10.3390/ijms22126410
Chicago/Turabian StyleSmirnov, Vasily, Olivier Grunewald, Jean Muller, Christina Zeitz, Carolin D. Obermaier, Aurore Devos, Valérie Pelletier, Béatrice Bocquet, Camille Andrieu, Jean-Louis Bacquet, and et al. 2021. "Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy" International Journal of Molecular Sciences 22, no. 12: 6410. https://doi.org/10.3390/ijms22126410
APA StyleSmirnov, V., Grunewald, O., Muller, J., Zeitz, C., Obermaier, C. D., Devos, A., Pelletier, V., Bocquet, B., Andrieu, C., Bacquet, J. -L., Lebredonchel, E., Mohand-Saïd, S., Defoort-Dhellemmes, S., Sahel, J. -A., Dollfus, H., Zanlonghi, X., Audo, I., Meunier, I., Boulanger-Scemama, E., & Dhaenens, C. -M. (2021). Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy. International Journal of Molecular Sciences, 22(12), 6410. https://doi.org/10.3390/ijms22126410